カルニチン
カルニチンは、脂肪酸の分解(β-酸化)を促進させる。
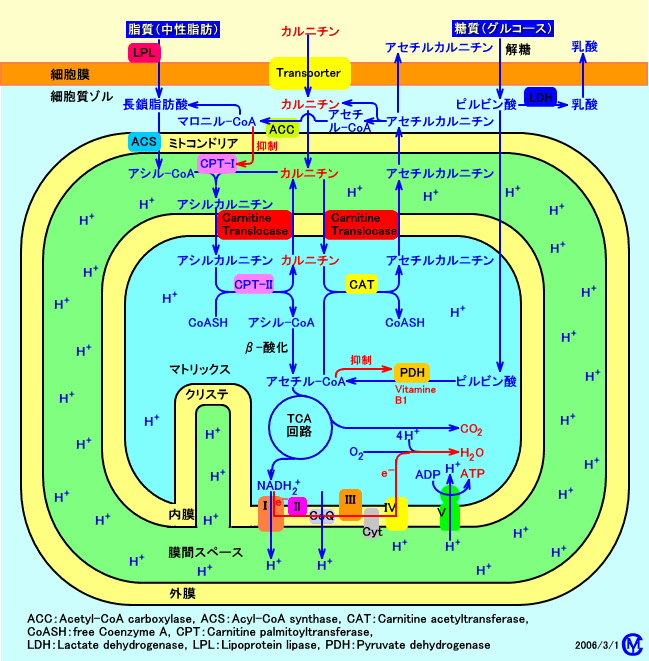
カルニチンは、長鎖脂肪酸をミトコンドリア内に輸送するのに、キャリアとして、必要。また、カルニチンは、ミトコンドリア内に蓄積したアシル-CoAを、アシルカルニチン(アセチルカルニチン)として、ミトコンドリア内に輸送するのに、必要。
1.カルニチンとは
カルニチン(carnitine)は、4-hydroxy-3-trimethylaminobutyric acid(β-hydroxy-γ-trimethyl ammonium butyrate:β-ヒドロキシ-γ-トリメチルアミノ酪酸)のこと。
化学式は、(CH3)3N+-CH2-CH(OH)-CH2-COOH。
古くは、ビタミンBTと呼ばれていた物質。
体内のカルニチンは、肝臓で生成されるか、食事から供給される。
カルニチンは、体内(肝臓、腎臓、脳)で、必須アミノ酸のリジンとメチオニンから、合成出来る:カルニチンは、肝臓で合成され、血液を介して、筋肉に取り込まれる(注1)。
カルニチンは、羊肉、赤肉、鶏肉、魚、乳製品などにも豊富に含まれていて、食事からも供給される。
植物は、カルニチンの生成に必要な2種類のアミノ酸(リジンとメチオニン)の含量が少ないので、ベジタリアンの人は、血漿カルニチン濃度や、尿中カルニチン排出量が、低下することがある。
カルニチン(アシルカルニチン)が欠乏すると、慢性疲労症候群と言う病気になるとも、言われる(注2)。
成人男性(体重70Kg)の体内には、カルニチンが約10mmol存在する。その体内カルニチンプールの内、98%は筋肉中に存在する。体内カルニチンプールの内、肝臓や腎臓に存在する量は、1.6%に過ぎない。体内カルニチンプールの内、0.6%は、細胞外液中に、遊離カルニチン、アシルカルニチン(カルニチンのβ位の水酸基に、脂肪酸がエステル結合している)として存在する。
2.カルニチンの役割
カルニチンは、脂肪酸(長鎖脂肪酸)を、β-酸化させる為に、ミトコンドリア内に輸送するのに必要。、
カルニチンは、ミトコンドリア内に蓄積した脂肪酸(アシル-CoA)を、ミトコンドリア外に輸送するのにも必要(ミトコンドリア内のCoA/アシル-CoA比が上昇し、遊離のCoAが増加する(遊離のCoAが供給される)。
a.カルニチンは、脂肪酸を、ミトコンドリア内に輸送するのに必要
カルニチンは、カルニチントランスポーターにより、細胞内に取り込まれる。
長鎖脂肪酸は、ミトコンドリア内で、β-酸化され、エネルギー源となる。
カルニチンは、長鎖脂肪酸のミトコンドリア内への輸送に必要。
細胞質の長鎖脂肪酸は、細胞質ゾルで、ACS(acyl-CoA synthetase)により、CoA(Coenzyme A)と結合して、アシル-CoA(acyl-CoA)に活性化される。ACS(acyl-CoA synthetase)は、acyl-CoA synthase、fatty acyl-CoA ligase、thiokinaseとも呼ばれる。
長鎖脂肪酸は、アシル-CoAとなった後、カルニチンと結合して、アシルカルニチン(Asylcarnitine)にならないと、ミトコンドリアの内膜を通過して、ミトコンドリア内のマトリックスに移行出来ない(注3)。なお、通常の食事を摂取している際には、健康人の血中に存在しない、中鎖脂肪酸(MCT)や、短鎖脂肪酸は、カルニチンと結合しなくても、ミトコンドリア内に輸送される。また、極長鎖脂肪酸(炭素数が26以上)は、主に、ペルオキシソーム(注4)で、分解される。
従って、カルニチンは、ミトコンドリア外の脂肪酸を、ミトコンドリア内に輸送するのに必須な物質(ビタミン)であり、カルニチンがないと、脂肪酸は、ミトコンドリア内のマトリックスで、β-酸化されにくい。
カルニチン(血液中の遊離カルニチン)が欠乏すると、脂肪酸(長鎖脂肪酸)のミトコンドリア内への輸送が低下し、脂肪酸のβ-酸化によるNADH2+等の生成が低下(更には、呼吸鎖によるATP生成も低下)し、組織の細胞内に、中性脂肪(トリグリセリド)が蓄積し、肝腫大や、ミオパチーが起こる。また、空腹時には、脂肪酸のβ-酸化による、NADH2+やATP等の生成が低下して、糖新生が低下して、低ケトン性低血糖を起こす。
b.カルニチンは、脂肪酸を、ミトコンドリア外に輸送するのに必要
ミトコンドリア内のアセチル-CoAは、カルニチンアセチルトランスフェラーゼ(CAT)の作用により、カルニチンと結合して、アセチルカルニチンとして、ミトコンドリア外の細胞質ゾルに、輸送される。アセチルカルニチンは、細胞質ゾルで、カルニチンとアセチル-CoAに分解され、アセチル-CoAは、アセチル-CoAカルボキシラーゼ(ACC)により、脂肪酸に合成される。
図には示さないが、カルニチンは、また、アシル-CoA(有害)が、ミトコンドリア内に蓄積した際に、カルニチンアシルトランスフェラーゼの作用により、アシル-CoAと結合し、アシルカルニチンとなり、細胞外に排泄される。
従って、カルニチンは、ミトコンドリア内のアセチル-CoAや、アシル-CoAを、ミトコンドリア外に輸送し、ミトコンドリア内のCoA/アシル-CoA比を調節する(遊離のCoAが増加する)のに必要な物質でもある。
3.全身性カルニチン欠損症
全身性カルニチン欠損症では、細胞膜のカルニチントランスポーターに障害(OCTN2遺伝子の異常)があり、骨格筋、心筋、腎臓で、血液中からカルニチンを、細胞内に取り込めない。また、血中カルニチンも減少している。細胞内カルニチンが欠乏するため、長鎖脂肪酸を、ミトコンドリア内へ転送する能力が低下する。そのため、脂肪酸は、ミトコンドリアでβ-酸化をされないため、飢餓などで、グルコースからアセチル-CoAを生成できない状況になると、エネルギー産生(ATP産生)が低下する。また、糖新生も低下して、ケトン体の上昇が少ない、低ケトン性低血糖を来たす。
また、中性脂肪(トリグリセリド)が組織に蓄積し、肝腫大(脂肪変性)、心筋障害、ミオパチーが起こる。
さらに、ミトコンドリア内に有害なアシル-CoAが蓄積し、ミトコンドリア内のCoA/アシル-CoA比が低下する(遊離のCoAが減少する)。その結果、TCA回路(ピルビン酸脱水素酵素)や、尿素回路(N-アセチルグルタミン酸合成酵素)が阻害され、エネルギー産生の低下、高乳酸血症、高アンモニア血症などを呈する( ライ症候群に類似した症状を来たす)。
カルニチン欠乏(カルニチン欠損)は、脂肪酸のβ-酸化に関与する酵素の欠損症(脂肪酸酸化異常症:FAOD)で、二次的に起こることの方が、多い。
4.CPT-I、CPT-II
CPT-I(carnitine O-palmitoyltransferase type I:カルニチンパルミトイルトランスフェラーゼ1型:EC 2.3.1.21)は、カルニチンと、脂肪酸(パルミチン酸:palmitic acid、palmitate))から生成されたアシル-CoA(palmitoyl-CoA)を結合させ、アシルカルニチン(palmitoylcarnitine)とする。
アシル-CoA+カルニチン→アシルカルニチン+CoASH
脂肪酸は、アシルカルニチンの形で、ミトコンドリアの内膜を通過して、ミトコンドリア内(マトリックス)に移行する(注4)。
CPT-I欠損症は、ライ様症候群を発症することがある。
CPT-II(carnitine O-palmitoyltransferase type II:カルニチンパルミトイルトランスフェラーゼ2型)は、ミトコンドリアのマトリックスで、アシルカルニチンを、カルニチンとアシル-CoAに分離する。
CPT-II欠損症は、乳児期に発症して、筋緊張低下、呼吸障害、不整脈、心肥大を来たす重症型と、成人期に発症して、発作性ミオグロビン尿症を来たすタイプがある。
CPT欠損症は、小児期に発症し、反復性の筋痛(筋肉痛)、筋腫脹、筋痙攣などの症状と、ミオグロビン尿が見られる(血液中のミオグロビン、CPKも、上昇する)。ミオグロビン尿の為、急性腎不全に陥る症例もある。
CPT欠損症の症状(発作)は、長時間の運動負荷、絶食、高脂肪食、寒冷などで、誘発される。
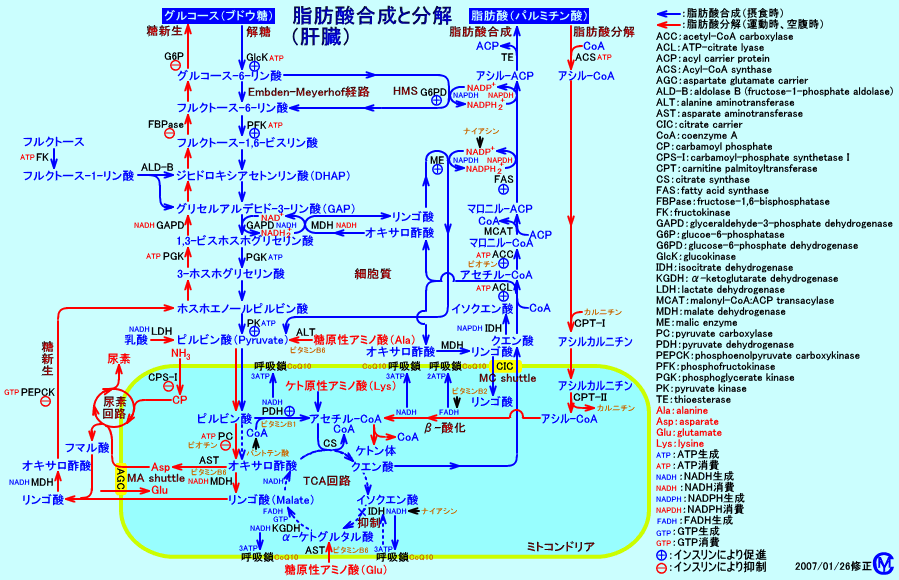
脂肪酸合成の中間代謝産物のマロニル-CoA(malonyl-CoA)は、CPT-1活性を抑制し、脂肪酸分解(脂肪酸のβ-酸化)を抑制する。
インスリンは、アセチル-CoAカルボキシラーゼ(ACC)の活性を促進し、脂肪酸合成を促進し、マロニル-CoA(malonyl-CoA)生成を促進する(脂肪酸分解を抑制する)。マロニル-CoAは、CPT-I(カルニチンアシルトランスフェラーゼI)の活性をアロステリックに阻害する。その結果、脂肪酸は、ミトコンドリア内への取り込み(脂肪酸分解)が抑制され、細胞質ゾルに留まる(インスリンは結果的にCPT-Iを抑制し脂肪酸のβ-酸化を抑制する)。
5.アセチル-CoAとアシル-CoAの代謝
a.アセチル-CoA
ミトコンドリア内のアセチル-CoAは、カルニチンアセチルトランスフェラーゼ(carnitine O-acetyltransferase:CAT.、注3)の作用により、カルニチンと結合して、アセチルカルニチンとして、ミトコンドリア外の細胞質ゾルに、汲み出される(ミトコンドリア内に、FreeなCoAが生成される)。
アセチル-CoA+カルニチン→アセチルカルニチン+CoASH
アセチルカルニチンは、細胞質ゾルから、血液中に排泄される。また、アセチルカルニチンは、細胞質ゾルで、カルニチンとアセチル-CoAに分解され、アセチル-CoAは、アセチル-CoAカルボキシラーゼ(acetyl-CoA carboxylase:ACC)により、マロニル-CoA経路で、脂肪酸に合成される。
b.アシル-CoA
虚血(低酸素)時には、長鎖脂肪酸ア シル-CoA(LCACoA)がミトコンドリア内に、蓄積する。LCACoAは、両親媒性(amphipathic)な性質の為、限界の濃度を超えると、ミトコンドリア膜に挿入され、膜の構造や、透過性(permeability)を変化させてしまう:パルミトイル-CoAの場合、30μMが、限界濃度。
図には示さないが、ミトコンドリア内にアシル-CoAが蓄積すると、カルニチンアシルトランスフェラーゼ(carnitine acyltransferase、注5)の作用により、カルニチンは、蓄積したアシル-CoAと結合して、アシルカルニチンとなる(ミトコンドリア内に、FreeなCoAが生成される)。
アシル-CoA+カルニチン→アシルカルニチン+CoASH
そして、ミトコンドリア内に蓄積したアシル-CoAは、アシルカルニチンとして、ミトコンドリア外(細胞質ゾル)に輸送され、血液中に排出される。
6.ミトコンドリア内での代謝
a.アセチルカルニチン
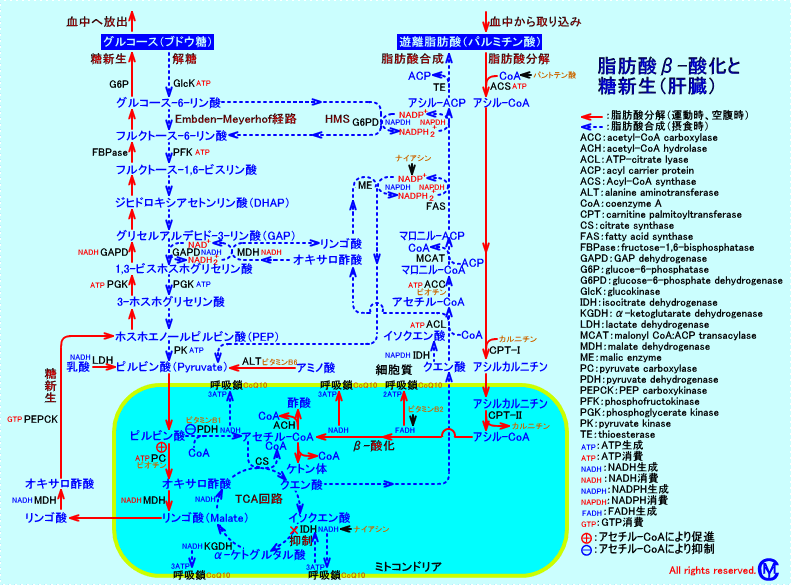
低酸素(hypoxia)などで、TCA回路(クエン酸回路)が障害されると、アセチル-CoAが代謝されないので、ピルビン酸の代謝物である乳酸が、血液中に増加する。
カルニチンは、CATにより、アセチル-CoAから、アセチルカルニチンを生成し、アセチルカルニチンとして、血中に輸送する。アセチル-CoAからアセチルカルニチンの生成は、ピルビン酸から乳酸の生成より行われ易いので、乳酸が減少する。
b.高脂肪食は、解糖を抑制する
脂肪酸のβ-酸化で生成されるアセチル-CoAは、ピルビン酸脱水素酵素を抑制する。
脂肪の摂取が多い(脂肪酸が多い)と、脂肪酸のβ-酸化で、アセチル-CoAが生成され、ピルビン酸脱水素酵素が抑制され、解糖(グルコースのピルビン酸への分解)を、抑制する。カルニチンは、脂肪酸のミトコンドリア内への輸送を促進し、脂肪酸のβ-酸化を促進するが、同時に、アセチル-CoAをアセチルカルニチンにして、アセチル-CoAによるピルビン酸脱水素酵素の抑制を、軽減する。
c.高炭水化物食は、脂肪酸のβ-酸化を抑制する
炭水化物の摂取が多いと、ピルビン酸脱水素酵素により、ピルビン酸からアセチル-CoAが生成され、アセチルカルニチンとして、細胞質ゾルに輸送され、アセチル-CoAから、アセチル-CoAカルボキシラーゼにより、マロニル-CoAが生成され、脂肪酸が合成される。マロニル-CoAは、CPT-Iを抑制し、脂肪酸のβ-酸化を抑制する。
6.抗生剤による、カルニチン欠乏
ピボキシル基を持つ抗生剤(CFPN-PIなど)は、消化管から吸収される際に、代謝を受け、抗菌活性物質と、ピバリン酸に分解される。ピバリン酸(ピボキシル基を有する抗生物質の代謝物)は、カルニチン抱合を受け、尿中にピバロイルカルニチンとして、排出される。従って、ピボキシル基を持つ抗生剤を長期間投与すると、血清中カルニチンが、低下する。
ピボキシル基を有する抗生剤(メイアクト、フロモックス、トミロンなど)を、通常量で、4〜5日間服薬すると、血清遊離カルニチン(濃度)は、30〜40%低下し、低カルニチン血症を来たすと言う。血清遊離カルニチンは、抗生剤の投与を中止すると、約2週間で、正常化する。低カルニチン血症により、脂肪酸酸化障害が起こると、低血糖により、痙攣、意識障害を来たすおそれがある。
2005年12月に、メイアクト(CDTR-PI)の添付文書が改訂され、「低カルニチン血症に伴う低血糖が、幼児に対してピボキシル基を有する抗生物質を長期投与した症例で報告されているので、痙攣、意識障害等の低血糖症状が認められた場合には投与を中止し、適切な処置を行うこと。」と、明記された。
低カルニチン血症による脂肪酸酸化障害により、低血糖を来たす理由は、まず、脂肪酸のβ-酸化により、NADH2+やアセチル-CoAが生成される。アセチル-CoAは、さらに、TCA回路で代謝されて、NADH2+が生成される。これらのNADH2+は、呼吸鎖で、ATP産生に利用されたり(運動時)、ATPと共に糖新生(飢餓時)に利用される。生体は、飢餓時など、グルコース(ブドウ糖)が不足した際に、肝臓で、糖新生を行い、グルコースを、血中に供給する。糖新生には、脂肪酸のβ-酸化により生成されるNADH2+やATPが必要なので、低カルニチン血症により、脂肪酸のβ-酸化が障害されると、糖新生が障害され、低血糖(低ケトン性低血糖)を来たす。
このように、感染症による発熱などによるストレスがかかったり、食事が摂取出来ない時(飢餓時)などに、抗生剤が原因で、低カルニチン血症による脂肪酸酸化障害を起こすと、肝臓では、糖新生に必要なNADH2+やATPが供給されない為、糖新生が行われず、低血糖(低ケトン性低血糖)を来たす。なお、脂肪酸のβ-酸化により、アセチル-CoAが生成されないと、糖新生に必要なピルビン酸カルボキシラーゼが活性化されない。また、筋肉では、脂肪酸のβ-酸化により、NADH2+やATPが生成されないので、グルコースの消費が高まることも、低血糖を来たす理由と言われる。
先天的な脂肪酸代謝異常症(脂肪酸酸化異常症:FAOD)でも、同様の機序により、低血糖を来たす。肝臓以外の組織(心筋や腎臓など9では、1分子の脂肪酸(パルミチン酸)が、カルニチンと結合し、ミトコンドリア内で、β-酸化されると、7分子のFADH2と7分子のNADH2+と、8分子のアセチル-CoAが生成され、129分子のATPが生成される:1分子のパルミチン酸(C16:0)が、β-酸化で1回分解され、1分子のアセチル-CoAが生成される際に、1分子のFADH2と1分子のNADH2+とが生じまる。ミトコンドリアの呼吸鎖で、FADH2やNADH2+から、それぞれ、2分子、及び、3分子のATPが生成される(計5分子のATPが生成される)。1分子のパルミチン酸は、アセチル-CoAに分解されるβ-酸化を7回繰り返すので、計5×7分子のATPが生成される(7回のサイクル数のβ-酸化により、8分子のアセチル-CoAが生成される)。アセチル-CoAが、TCA回路で代謝されると、11分子のATPと1分子のGTPが生成される。従って、β-酸化とTCA回路で、計35+(12×8)=131分子のATPに相当するエネルギーが生成される。しかし、パルミチン酸(C16:0)が、β-酸化される初期の段階で、Thiokinase(ACS)により、アシル-CoA(C15-CO-CoA)に変化させるのに、1分子のATPが消費されAMPになる(実質、2分子のATPに相当するエネルギーが消費される)ので、差し引き、129分子のATPが生成される。
肝臓でも、1分子のパルミチン酸がβ-酸化されると、7分子のFADH2と7分子のNADH2+と、8分子のアセチル-CoAが生成される。しかし、肝臓では、パルミチン酸のβ-酸化により生成された8分子のアセチル-CoAは、TCA回路に入らず、ケトン体のアセト酢酸になる。従って、肝臓では、1分子のパルミチン酸がβ-酸化されると、差し引き、(2×7)+(3×7)−2=33分子のATPが、生成される。
1分子(1mol)のグルコース(ブドウ糖)の代謝では、38分子のATP(グルコース100g当り21分子のATP)が生成される。1分子(1mol)のパルミチン酸の代謝では、肝臓以外の組織では、129個分子のATP(パルミチン酸100g当り50分子のATP)が生成される。従って、脂肪酸の方が、ブドウ糖より、エネルギー力価が高い(ATPは、100g当たり、脂肪酸の方が、29分子多く、生成される)。
7.カルニチンの代謝
カルニチンの体内総プール量は、100mmolであり、その内、98%が筋肉内に、1.6%が肝臓と腎臓に、0.6%が細胞外液中に存在する。
カルニチンは、大半は、尿中(腎臓)から排泄され、1%以下は、便中から、排泄される。腎臓は、体内では分解されないカルニチンを排泄し、カルニチンの体内貯蔵量を調節している。
腎臓でのクリアランスは、アシルカルニチンは、遊離カルニチンより、10〜20倍速い。アセチルカルニチンは、腎尿細管から再吸収されるが、長鎖脂肪酸のアシルカルニチンは、再吸収が悪い。その為、脂肪酸代謝異常症(脂肪酸酸化異常症:FAOD)では、代謝が阻害された脂肪酸の炭素鎖が長い程、アシルカルニチンとして、カルニチンが、尿中に排泄されるので、血中のカルニチンは、減少し易い。
尿中に排泄される、アシルカルニチンを分析することにより、脂肪酸代謝異常症(脂肪酸酸化異常症:FAOD)や、有機酸代謝異常症を、診断することが可能。
ライ症候群でも、ミトコンドリアの脂肪酸代謝(β-酸化)が障害され、アシルカルニチンが、尿中に増加する。正常児のアシルカルニチンのアシル基は、アセチル基(アセチール)である(アセチルカルニチン)が、ライ症候群のアシルカルニチンのアシル基は、アシル基とされる。また、ライ症候群に類似した症状を示す、カルニチン欠損症と異なり、ライ症候群では、血中カルニチン値は、典型的には、低下しないが、二次的に低下することがある。
ライ症候群やライ様症候群(Reye症候群類似疾患)では、多くの場合、アシル-CoAが蓄積する。
ライ症候群では、肝組織中に、短鎖アシル-CoA、中鎖アシル-CoA、分岐鎖アシル-CoAが蓄積する:octanoyl-CoA、isovaleryl-CoA、butyryl-CoA、isobutyryl-CoA、propionyl-CoA、methylmalonyl-CoAが蓄積する(中鎖脂肪酸や、短鎖脂肪酸は、カルニチンと結合しなくても、ミトコンドリア内に輸送されるので、カルニチンはキャリアとして重要でない)。
ライ症候群では、肝組織中のアセチル-CoAは、正常の半分程度の値が、存在する。ライ症候群では、(肝組織中の)遊離CoAは、著明に減少し(正常の10%)、遊離CoA/アセチル-CoA比は、0.,5以下になる。
FAODの一つ、中鎖アシル-CoA脱水素酵素欠損症(MCADD)では、肝組織中のアセチル-CoAは、著明に減少する点が、ライ症候群と異なる。
酵素放射性同位元素法(ERI法)により測定した小児の正常値は、血清総カルニチン61.8±13.1nmol/ml、遊離カルニチン45.6±11.0nmol/ml、アシルカルニチン16.2±7.6nmol/mlで、アシルカルニチン/総カルニチン比は、26.0±10.0%とされる。
血清総カルニチン値が低値の場合は、組織のカルニチンも、欠乏していることが多い。
アシルカルニチン/総カルニチン比は、カルニチン代謝の異常を反映する:何らかの代謝異常により、組織のミトコンドリア内に、アシル-CoAが過剰に酸性され蓄積すると、アシルカルニチンの生成が促進され、血中に輸送され、アシルカルニチン/総カルニチン比が上昇する。
新生児早期は、(肝臓での)カルニチン生成能が低い為、カルニチンを含まないミルクで育てると、容易にカルニチン欠乏(低カルニチン血症)に陥る。
8.CoA/アシル-CoA比の調節
CoA量(細胞内CoA量とミトコンドリア内CoA量)には、限りがある。
脂肪酸のβ-酸化が、最大速度で行われると、CoAは、結合型CoA(アシル-CoAやアセチル-CoA)となり、遊離型CoA量が、不足して来て、β-酸化が抑制される。
アセチル-CoAヒドロラーゼ(acetyl-CoA hydrolase:EC 3.1.2.1、アセチル-CoAヒドラーゼ)は、ミトコンドリア内で、遊離型CoAが不足すると、アセチル-CoAを加水分解し、遊離型CoAと、酢酸を生成する。アセチル-CoAヒドラーゼの活性は、遊離型CoAにより、阻害される。
注1:蛋白質のリジン残基が、メチル化され、トリメチルリジンとなり、加水分解され遊離され、肝臓、心筋、骨格筋、腎臓などで、カルニチン前駆体のγ-ブチロベタインが、合成される。γ-ブチロベタインは、最終的に、肝臓、腎臓、脳に存在する、γ-ブチロベタインヒドロキシラーゼにより、カルニチンに合成される。γ-ブチロベタインヒドロキシラーゼは、肝臓、腎臓、脳にのみ存在し、筋肉(骨格筋、心筋)には存在しない。
肝臓などで合成されたカルニチンは、血中を運ばれ、末梢細胞(筋細胞など)で、細胞膜のナトリウム依存性トランスポートシステム(Transporter)により、細胞内に取り込まれる(能動輸送)。細胞内カルニチン濃度は、細胞外液より、20〜50倍、高く維持されている。
注2:慢性疲労症候群(CFS)は、風邪などを契機に、激しい倦怠感や、脱力感を、半年以上、持続ないし繰り返す病気で、病因は、確定されていない。
ACR(アシルカルニチン)は、疲労の程度と相関している。ACRは、グルタミン酸、アスパラギン酸、GABA(γアミノ酪酸)などの神経伝達物質の合成に利用される。慢性疲労症候群では、ACR(アシルカルニチン)の脳内(前頭葉の前帯状回と、前頭前野)への取り込みが低下していると言わる。慢性疲労症候群では、アセチルカルニチンの脳内への取り込みが、Broadmann24野(自律神経系の調節や情動などに関連する)と、Broadmann9野(意欲やコミュニケーションに関連する)において、局所脳血液量低下とは無関係に、低下していると言われる。アシルカルニチンや、アセチルカルニチンが低下しているのに、脳内への取り込みも低下していることは、慢性疲労症候群では、ストレス(メルトダウン)により、体内の代謝が抑制され、アシルカルニチンや、アセチルカルニチンの、産生(合成)や、細胞内への取り込みがしていることを示唆しているのかも知れない。
慢性疲労症候群では、血清コルチゾールや、DHEA-S(デヒドロエピアンドロステロンサルフェート)など、副腎皮質から産生されるホルモンが減少し、思考力や集中力が低下すると言われる。DHEA-Sは、アセチルカルニチントランスフェラーゼ(acetylcarnitine transferase:carnitine O-acetyltransferase:CAT)活性と相関していて、DHEA-Sが減少すると、アセチルカルニチンが低下する。DHEA-Sを投与(補充)すると、アセチルカルニチンが上昇する。この説では、アセチルカルニチンの低下は、慢性疲労症候群の原因ではなく、結果を意味する。ストレスにより、内分泌系が崩れ、DHEA-Sなどのホルモン産生のバランスが、失調することが、慢性疲労症候群の第一原因なのかも知れない。
慢性疲労症候群では、NK細胞活性が低下し、潜伏感染していたウイルス(HHV-6、EBVなど)が、再活性化すると言われる。また、IFN-α(インターフェロンアルファ)は、2,5-AS(2',5'-オリゴアデニル酸合成酵素:2,5-oligoadenylate synthetase)を誘導し、誘導された2-5A(2',5'-オリゴアデニル酸)により、RNaseを活性化させ、ウイルスRNAを分解して、ウイルス増殖を抑制するが、慢性疲労症候群では、2-5ASが上昇している。しかし、HHV-6、EBVなどのウイルスは、初感染後、体内に、潜伏感染するウイルスであり、特に、HHV-6は、過労状態の人では、高率に唾液中に分泌(検出)される。従って、ウイルスの再活性化や、2-5ASの上昇は、慢性疲労症候群の原因と捉えるより、ストレスにより、免疫力が低下(疲弊)した結果と、捉える方が、妥当と思われる。
ウイルスの再活性化に伴い、抗炎症性サイトカインのTGF-βが増加する。TGF-βは、DHEAの硫酸抱合を調節している酵素(スルフォキナーゼ)の活性を抑制する為、DHEA-Sが減少し、アセチルカルニチンの合成が低下する。
慢性疲労症候群では、過度なストレスから生体機能を守る為に、生体の生理的な安全機構(自己防衛機制)が作動し、コルチゾールやDHEA-Sの産生を抑制したり、脳血流を低下させて脳代謝を抑制しているのかも知れない。
なお、アンドロゲン(男性ホルモン)には、睾丸から分泌されるテストステロンと、副腎から分泌されるDHEA-S(デヒドロエピアンドロステロンサルフェート)とがある。アンドロゲンは、男性ホルモンの総称で、最も重要なテストステロンは、主に睾丸(精巣)で作られて、分泌され、末梢組織で、5α還元酵素により、活性型の5α-DHT(デヒドロテストス
テロン)に変換される。この5α-DHTの受容体が欠損すると、染色体は男性型で、睾丸もあるのに、体系は女性型になる(睾丸性女性化症)。
注3:アシルカルニチンは、porinと言うミトコンドリア外膜の穴を通過して、ミトコンドリア内に、輸送されるとも言われている。
肝臓では、遊離脂肪酸から生成されるアシル-CoAが、カルニチンシャトルにより、ミトコンドリア内に取り込まれ、アセチル-CoAとなり、TCA回路で代謝される。インスリンは、このアシル-CoAのミトコンドリア内への取り込みを、抑制する。インスリン不足・インスリン拮抗ホルモン過剰の際には、アシル-CoAのミトコンドリアへの取り込みが促進され、大量のアシル-CoA(遊離脂肪酸から生成される)が、ミトコンドリア内に流入し、TCA回路で代謝されないと、アセチル-CoAから、ケトン体が生成され、血中ケトン体が増加し、ケトーシスを来たす。
注4:ペルオキシソーム(peroxisome)は、細胞内小器官の一つ。
生物学的には、長鎖脂肪酸のβ-酸化は、本来、ミトコンドリアでなく、ペルオキシソームで行われていた。
ペルオキシソーム(peroxisome)は、peroxide(過酸化水素:H2O2)と、some(顆粒)とから命名されている。
ペルオキシソームでは、過酸化水素(H2O2)の生成と分解が行われるので、種々のオキシダーゼやカタラーゼが存在している。
ペルオキシソームでは、極長鎖脂肪酸のβ-酸化(長鎖脂肪酸のβ-酸化はミトコンドリアで行われる)や、エーテルリン脂質の生成なども行われる。
注5:カルニチンアシルトランスフェラーゼ(EC 2.3.1.)は、ミトコンドリア内膜に存在し、活性脂肪酸(acyl-CoA)のアシル基(acyl groups)を、カルニチン(L-carnitine)に転移させ、ACR(アシルカルニチン)を生成することで、ミトコンドリア内のacyl-CoAを、ミトコンドリア内膜を経て、ミトコンドリア外に輸送させる。
・カルニチンアセチルトランスフェラーゼ(carnitine O-acetyltransferase:CAT:EC 2.3.1.7.)は、カルニチンアシルトランスフェラーゼ(EC 2.3.1.)であり、acetyl-CoA(C2)以外に、propanoyl-CoA(C3)や、butanoyl-CoA(C4)を、カルニチンと結合させ、アセチルカルニチンやアシルカルニチンを生成する。
acetyl-CoA + carnitine = CoA + O-acetylcarnitine
・カルニチンパルミトイルトランスフェラーゼ(carnitine O-palmitoyltransferase:CPT:EC 2.3.1.21.)も、アシルトランスフェラーゼであり、炭素数が8〜18(C8〜C18)の、広い範囲の脂肪酸に作用するが、パルミチン酸(C16)の活性脂肪酸(palmitoyl-CoA)に、最も作用する。
palmitoyl-CoA + L-carnitine = CoA + L-palmitoylcarnitine
・カルニチンオクタノリルトランスフェラーゼ(carnitine O-octanoyltransferase:EC 2.3.1.137.)は、中鎖〜長鎖(medium-chain/long-chain)の活性脂肪酸(acyl-CoAs)に作用し、特に、炭素数が6〜8(C6〜C8)のアシル基(acyl groups)に、例えば、オクタン酸(C8)の活性脂肪酸(octanoyl-CoA)を始めとして、作用する。
octanoyl-CoA + L-carnitine = CoA + L-octanoylcarnitine
参考文献
・ハーパー・生化学(原著14版、三浦義彰監訳、丸善株式会社、 1975年).
・ヴォート基礎生化学(東京化学同人、第1版第4刷、2003年).
・鈴木紘一、他:ホートン生化学 第3版(東京化学同人、2005年、第3刷).
・シンプル生化学(改訂4版、南江堂、2003年).
・小児科 40巻8号、1042〜1048頁、1999年.
・大浦敏博:全身性カルニチン欠損症とカルニチン療法 小児科 Vol.40 No.9、1042-1048、1999年.
・大浦敏博:カルニチン欠乏症と補充療法 小児科 Vol.34 No.11、1377-1385、1993年.
・山田研太郎:急性合併症のための検査と処置 糖尿病診療マニュアル 日本医師会雑誌 特別号 Vol.130 No.8、S230-233、 2003年.
・山本重則、他:ミトコンドリア脂肪酸酸化異常症 36: 1293-1299, 1995年.
・田川邦夫:からだの働きからみる代謝の栄養学 タカラバイオ株式会社(2003年).
・富永昌子、他:カルニチンパルミトイル転位酵素I欠損症の1女児例 日本小児科学会雑誌 107巻6号、917-921、2003年.
・浅井清文、和田義郎:カルニチン代謝異常 小児科 Vol.30 No.4、363-368、1989年.
・佐久間徹、他:カルニチン療法 小児内科 Vol.23 No.12、1863-1869、1991年.
・吉田一郎、松石豊次郎:Reye症候群類似疾患とカルニチン 小児科 Vol.30 No.9、1023-1028、1989年.
・豊田隆謙:特集 インスリンをめぐる諸問題 インスリン代謝、日本医師会雑誌、第112巻・第1号、27-31頁、平成6年7月1日号.