ミトコンドリア脂肪酸酸化異常症(FAOD)
【ポイント】
脂質(脂肪酸)は、体内で、糖(グルコース)に変換出来ないので、脂肪酸の代謝異常は、直接的には、血糖値に影響しない。
しかし、糖新生には、脂肪酸が、ミトコンドリア内でβ-酸化されて生成されるNADH2+や、β-酸化で生成されたアセチル-CoAがTCA回路や呼吸鎖で代謝されて生成されるNADH2+やATPが必要なので、脂肪酸の代謝異常や、ミトコンドリアの機能障害では、糖新生が障害され、低ケトン性低血糖症を来たす。
ミトコンドリア脂肪酸酸化異常症(FAOD)では、脂肪酸はミトコンドリアに入ってβ-酸化されないので、ケトン体が生成されない為、低ケトン性低血糖症を来たす。また、FAODで、脂肪酸が分解(β-酸化)されないと、各臓器に、脂肪(中性脂肪)が蓄積し、脂肪変性が生じる。
1.FAOD
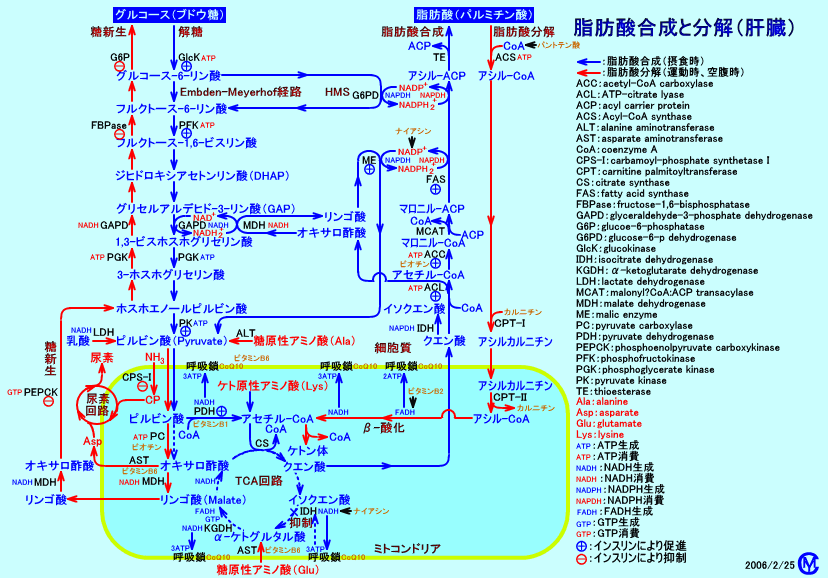
ミトコンドリアやペルオキシソームには、脂肪酸をβ-酸化させる脂肪酸酸化経路が存在する。
ミトコンドリアの脂肪酸酸化経路を構成する、酵素蛋白質、転送蛋白質などが、欠損すると、ミトコンドリア脂肪酸酸化異常症(mitochondrial fatty acid oxidation defect:FAOD)を来たす。
FAODは、肝機能障害を伴うライ症候群類似(mimicker of Reye syndrome)の急性脳症を発症したり、乳幼児突然死症候群(SIDS:sudden infant death syndrome)の原因となることがある。
FAODでは、感染症や飢餓の際に、低ケトン性低血糖が見られる。
FAODは、周期性嘔吐症(アセトン血性嘔吐症、自家中毒)とし、誤診されることもある。
FAODは、脂肪酸が分解(β-酸化)されないので、飢餓時など、グルコース(ブドウ糖)からエネルギー(ATP)を産生出来なくなった際に、肝臓や筋肉などで、脂肪酸からエネルギー(ATP)を産生したり、糖新生が行われない(糖新生に必要なNADH2+等が供給されない)為、低ケトン性低血糖を来たす。
FAODで、脂肪酸が分解(β-酸化)されないと、各臓器に、脂肪(中性脂肪)が蓄積し、脂肪変性が生じる。心筋の脂肪変性により、心筋障害、心肥大が、骨格筋の脂肪変性により、筋力低下、筋緊張低下、横紋筋融解症、脂肪肝などを、来たす。
FAODには、以下のような欠損症が存在する。
1).アシルCoA生成の異常
・カルニチンパルミトイルトランスフェラーゼ1型欠損症:CPT-I欠損症(carnitine palmitoyltransferase 1 deficiency)
・カルニチンパルミトイルトランスフェラーゼ2型欠損症:CPT-II欠損症(carnitine palmitoyltransferase 2 deficiency)
・原発性カルニチン欠損症
・カルニチン転位酵素欠損症
・トランスロカーゼ欠損症
2).ミトコンロドリア内膜長鎖脂肪酸酸化の異常
・極長鎖アシル-CoA脱水素酵素欠損症:VLCAD欠損症(very long-chain acyl-CoA dehydrogenase deficiency)
・長鎖3-ヒドロキシルアシル-CoA脱水素酵素欠損症(LCHADD)
・長鎖アシル-CoA脱水素酵素欠損症:LCAD欠損症(long-chain acyl-CoA dehydrogenase deficiency)
3).ミトコンドリアマトリックス中鎖・短鎖脂肪酸酸化の異常
・中鎖アシル-CoA脱水素酵素欠損症:MCAD欠損症(medium-chain acyl-CoA dehydrogenase deficiency:MCADD)
・短鎖アシル-CoA脱水素酵素欠損症:SCAD欠損症(short-chain acyl-CoA dehydrogenase deficiency:SCADD)
・短鎖3-ヒドロキシアシル-CoA脱水素酵素欠損症(SCHADD)
・中鎖3-ケトアシルCoAチオラーゼ欠損症(MCKATD)
・短鎖3-ケトアシルCoAチオラーゼ欠損症(SCKATD)
4).電子伝達系への橋渡しの異常
・グルタル酸血症II型(GAII:グルタル酸尿症2型):ETF欠損症(ETF欠損症、注1)、ETF脱水素酵素欠損症(ETF-DH欠損症)
5).ケトン体の生成の異常
・3-ヒドロキシ-3-メチルグルタリルCoAリアーゼ欠損症(HMGLD)
・HMG合成酵素欠損症(HMGSD)
ミトコンドリアでは、脂肪酸をβ-酸化により分解し、エネルギー(ATP)を供給する。
脂肪酸は、肝臓、心筋、骨格筋では、主要なエネルギー源とされている。
特に、空腹時、激しい運動時、感染時など、ブドウ糖(グルコース)を解糖してエネルギーを十分に供給できない時に、主に、脂肪酸をエネルギー源とするので、ミトコンドリアの脂肪酸β-酸化系は、重要な役割を果たす。
そのため、FAODは、空腹時、激しい運動時、感染時などに、ブドウ糖からのエネルギー供給が不足した時に、急性発症しやすい。
特に、肝臓、骨格筋、心筋は、β-酸化が盛んな臓器であり、FAODの異常所見が出易い。
FAODの異常所見としては、肝腫大、脂肪肝、筋緊張低下、筋肉痛、心拡大、心筋障害、伝導障害などが起こり得る。
FAODの検査所見としては、低血糖、血液中のAST(GOT)、LDH、CK(CPK)、FFA(遊離脂肪酸)などの上昇や、高アンモニア血症、代謝性アシドーシスなどが見られる。原因不明の高CK血症(高CPK血症)の原因として、ミトコンドリア脂肪酸β-酸化異常症(FAOD)が、見つかることがある。
FAODの多くの患者は、安定期には無症状だが、間歇的に発作を起こし、2歳未満(30/39例)に、発症する。
初発症状としては、急性脳症を発症することが少なくなく(21/39例)、肝機能障害を伴うライ症候群類似(mimicker of Reye's syndrome)の症状を示すことがある。
乳幼児突然死症候群(SIDS:2/39例)を来たしたり、筋緊張低下や筋肉痛などの骨格筋症状(23/39例)、発達遅滞も見られる。肝機能障害(34/39例)、低血糖(14/28例)、高アンモニア血症(18/22例)が見られる。
FAODでは、脂肪酸はミトコンドリアに入ってβ-酸化されず、ケトン体は生成されない。
FAODの乳児は、低血糖があるのに、尿ケトン体の濃度が低いことが、検査所見の特徴(低ケトン性低血糖)。
CPT-II欠損症は、乳児期に発症して、筋緊張低下、呼吸障害、不整脈、心肥大を来たす重症型と、成人期に発症して、発作性ミオグロビン尿症を来たすタイプがある。
VLCAD欠損症は、乳児期(生後3〜4カ月)に発症して、肥大型心筋症、筋緊張低下、肝機能障害、低血糖を主徴とするタイプと、青年期に運動や空腹時に発症して、横紋筋融解症を主徴とするタイプがある。
LCAD欠損症は、乳児期より、心筋症、筋緊張低下、肝腫大、低血糖を来たす。
原発性全身性カルニチン欠損症(primary systemic carnitine deficiency:PCD)では、進行性の心筋症(拡張性心筋症)を、1〜7歳頃に発症する。また、3カ月〜2.5歳頃に、低ケトン性低血糖症を呈する、急性脳症で発症することもある。肝生検では、脂肪変性があり、ライ症候群と診断されることも多い。血中のカルニチン濃度は、正常の10%以下の5μmol/L以下に低下する。カルニチンを経口投与すると、症状は劇的に改善する。
全身性カルニチン欠損症は、カルニチントランスポーター遺伝子(OCTN2遺伝子)に異常がある。細胞膜のカルニチントランスポーターに障害があり、カルニチンを、骨格筋、心筋、腎で、血液中から細胞内に転送(能動輸送)出来ない(腎臓で再吸収されないため、カルニチン血中濃度も低下する)。肝臓では、カルニチンを細胞内に転送出来る。
短鎖アシル-CoA脱水素酵素欠損症では、ブチリルCoA(炭素数4)のβ-酸化は、障害されるが、炭素数6以上のアシルCoAのβ-酸化は、それ程、障害されず、エネルギー産生の低下症状は、強くない。
カルニチンパルミトイルトランスフェラーゼ1型欠損症(CPT-I欠損症、カルニチンパルミトイルトランスフェラーゼI型異常症)を除いて、FAODでは、一般に、血清中のフリーカルニチンは低下し(二次性カルニチン欠乏症)、アシルカルニチン/フリーカルニチン比が、増加する。
長鎖アシルカルニチンは、CPT-II欠損症(肝型と筋型のCPT-II異常症)、VLCAD欠損症(VLCAD異常症)、LCAD欠損症(LCAD異常症)、Translocase異常症で、血清中に増加する。
中鎖アシルカルニチンは、MCAD欠損症(MCAD異常症)で、血清中に増加する。
2、ライ症候群とFAOD(ライ様症候群)
FAODは、ライ様症候群を来たす。
ライ症候群やライ様症候群(Reye症候群類似疾患)では、多くの場合、アシル-CoAが蓄積する。
ライ症候群では、肝組織中に、短鎖アシル-CoA、中鎖アシル-CoA、分岐鎖アシル-CoAが蓄積する:octanoyl-CoA、isovaleryl-CoA、butyryl-CoA、isobutyryl-CoA、propionyl-CoA、methylmalonyl-CoAが蓄積する(中鎖脂肪酸や、短鎖脂肪酸は、カルニチンと結合しなくても、ミトコンドリア内に輸送されるので、カルニチンはキャリアとして重要でない)。
ライ症候群では、肝組織中のアセチル-CoAは、正常の半分程度の値が、存在する。ライ症候群では、(肝組織中の)遊離CoAは、著明に減少し(正常の10%)、遊離CoA/アセチル-CoA比は、0.,5以下になる。
FAODの一つ、中鎖アシル-CoA脱水素酵素欠損症(MCADD)では、肝組織中のアセチル-CoAは、著明に減少する点が、ライ症候群と異なる。
ライ症候群では、ミトコンドリア脂肪酸β-酸化異常の結果、尿中アシルカルニチン分析では、アセチルカルニチンのほかに、アシルカルニチン(C6〜C12のジカルボン酸)が検出される。
ライ症候群では、ミトコンドリア障害の為、ミトコンドリア内で、長鎖脂肪酸(アシル-CoA)の代謝で生成されるアセチルカルニチンや、中鎖脂肪酸や短鎖脂肪酸の代謝で生成されるアシルカルニチン(C6〜C12のジカルボン酸に対応するC6〜C12 dicarboxylic acylcarnitine)が、尿中に増加する。(尿中の)C6〜C10 dicarboxylic acylcarnitineは、C6〜C10 dicarboxylic acidの10%以下だが、C12 dodecandionylcarnitineは、C12 dodecandioic acidより高濃度。ライ症候群では、ジカルボン酸のβ-酸化が一部障害され、dodecandionyl-CoAが蓄積し、dodecandionylcarnitineが生成されると言う。
カルニチンは、長鎖脂肪酸をミトコンドリア内に輸送するのに、キャリアとして、必要。
FAODでは、ジカルボン酸が、尿中に、多量に排泄される(ジカルボン酸尿症)。FAODでは、低ケトン性ジカルボン酸尿症で、セバシン酸(C10)/アジピン酸(C6)比が、高い。ジカルボン酸尿症は、アセトン血性嘔吐症や、飢餓時など、脂肪酸酸化が亢進した状態でも、認められるが、これらの場合は、ケトン性ジカルボン酸尿症であり、ケトン体合成が、亢進する。ジカルボン酸尿症は、MCTミルクや、MCTオイルなど、中鎖トリグリセリド(中性脂肪)を多量に摂取した場合にも、認められるが、この場合も、ケトン性ジカルボン酸尿症であり、ケトン体合成が、亢進する。
3.その他
・脂肪酸酸化異常症(FAOD)を、新生児時期に、マススクリーングで発見する際、充分にカロリーが補給され、脂肪酸β-酸化系に依存しない代謝状況になっていると、指標値が、異常を示さない場合がある。
・食事中の中鎖脂肪酸(MCT:Medium Chain Triglycerides)は、腸管内腔から門脈を経て吸収される(リンパ管を経ずに肝臓に輸送されるので、吸収・分解の効率が良いと言われる)。
長鎖脂肪酸(LCT:Long Chain Triglycerides)は、小腸で吸収された後、再エステル化され、カイロミクロンとなって、リンパ管(胸管)を経て大循環(大動脈)に入り、肝臓に輸送される。
MCTは、LCTと異なり、ミトコンドリア内にβ-酸化される為に取り込まれる際に、カルニチンを必要としない(MCTの方が代謝される速度がLCTの5倍速い)。
表1 脂肪酸の分類(参考文献の目黒英二氏の表2を改変し引用) 短鎖脂肪酸 中鎖脂肪酸(MCT) 長鎖脂肪酸(LCT) 飽和脂肪酸 酪酸(C4:0)
ヘキサン酸(カプロン酸:C6:0)
オクタン酸(カプリル酸:C8:0)
デカン酸(カプリン酸:C10:0)
ラウリン酸(C12:0)ミリスチン酸(C14:0)
パルミチン酸(C16:0)
ステアリン酸(C18:0)
不飽和脂肪酸 オレイン酸(C18:1)
リノール酸(C18:2)
α-リノレン酸(C18:3)
γ-リノレン酸(C18:3)
アラキドン酸(C20:4)
エイコサペンタエン酸(EPA:C20:5)
ドコサヘキサエン酸(DHA:C22:6)
・新生児時期に先天的な代謝異常をスクリーニングにする方法として、従来から、ガスリー法が用いられて来た。ガスリー法では、フェニルケトン尿症、ホモシスチン尿症、メープルシロップ尿症、クレチン症(先天性甲状腺機能低下症)、先天性副腎過形成、ガラクトース血症を検出出来た。
タンデムマスと言う質量分析計により検査(タンデムマススクリーニング)すると、一挙に多種類の先天性代謝異常症の有無を調べることが可能。
タンデムマスを用いると、以下のような代謝異常症を検出出来る。
・アミノ酸代謝異常症:フェニルケトン尿症、メープルシロップ尿症、ホモシスチン尿症、高チロシン血症I型
・有機酸代謝異常:メチルマロン酸血症(MMA)、プロピオン酸血症(PA)、イソ吉草酸血症(IVA)、3-メチルクロチニルグリシン尿症(MCC)、グルタル酸血症1型(GAI:グルタル酸尿症1型)、3-ヒドロキシ-3-メチルグルタル酸尿症(HMG血症)、複合カルボキシラーゼ欠損症(マルチプルカルボキシラーゼ欠損症:MCD)、βケトチオラーゼ欠損症
・脂肪酸代謝異常:短鎖3-ヒドロキシアシル-CoA脱水素酵素欠損症(SCHADD)、中鎖アシルCoA脱水素酵素欠損症(MCAD欠損症)、極長鎖アシルCoA脱水素酵素欠損症(VLCAD欠損症)、三頭酵素/長鎖3-ヒドロキシアシルCoA脱水素酵素欠損症(TFP/LCHAD欠損症)、カルニチンパルミトイルトランスフェラーゼ-1欠損症(CPT-I欠損症)、カルニチンパルミトイルトランスフェラーゼ-2欠損症(CPT-II欠損症)、カルニチンアシルカルニチントランスロカーゼ欠損症(CACT欠損症:TRANS)、全身性カルニチン欠乏症(カルニチントランスポータ異常症)、グルタル酸血症2型(GAII:グルタル酸尿症2型:マルチプルアシルCo-A脱水素酵素欠損症)
・尿素回路異常症:シトルリン血症I型(古典型:アルギニノコハク酸合成酵素欠損症)、アルギニノコハク酸尿症(アルギニノコハク酸リアーゼ欠損症)、高アルギニン血症(アルギナーゼ欠損症)
・その他:シトリン欠損症(CTLN2)
タンデムマスでは、ガスリー法で検出出来る、クレチン症(先天性甲状腺機能低下症)、先天性副腎過形成、ガラクトース血症は、検出出来ない。ガスリー法の検査に用いる新生児の血液を染み込ませた濾紙は、タンデムマスの検査に用いることが出来る。
新生児(約127万人)をタンデムマスで検査した結果、約9,000人に1人の頻度で、代謝異常が発見されたと言う。
注1:ETFとは、electron transfer flavoprotein(電子伝達フラビン蛋白)の略。ETFは、脂肪酸のβ-酸化で生成されるFADH2から、電子を電子伝達系に伝達する。
参考文献
・小児科 40巻8号、1042〜1048頁、1999年.
・小児科 40巻13号、1743〜1751頁、1999年.
・日本小児科学会雑誌 102巻7号、753〜758頁、1998年.
・山本重則、他:ミトコンドリア脂肪酸酸化異常症 36: 1293-1299, 1995年.
・吉田一郎、松石豊次郎:Reye症候群類似疾患とカルニチン 小児科 Vol.30 No.9、1023-1028、1989年.
・大竹明、他:脂肪酸β酸化異常症−その臨床像の特徴と遺伝子解析の現況− 小児科Vol.32 No.3、249-254、1991年.
・大浦敏博:全身性カルニチン欠損症とカルニチン療法 小児科 Vol.40 No.9、1042-1048、1999年.
・大浦敏博:カルニチン欠乏症と補充療法 小児科 Vol.34 No.11、1377-1385、1993年.
・重松陽介:タンデム質量分析計による新生児代謝異常症マススクリーニング、日本小児科学会雑誌、110巻7号、895-903、2006年.
・目黒英二:Q37 投与する脂質にはどんなものがあるの?、ナーシングQ&A 全科に必要な栄養管理Q&A、80-81頁、総合医学社(2005年).
・新生児の先天的代謝異常症 新検査法導入段階に 「タンデムマス」20以上の疾患判定、信濃毎日新聞、2011年8月26日金曜日、13面.