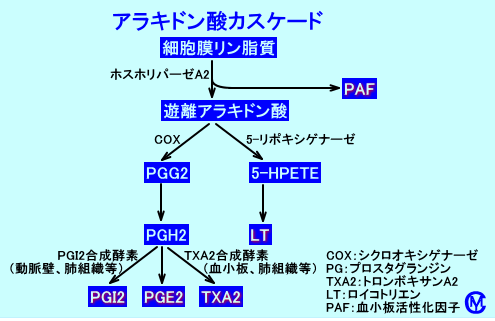
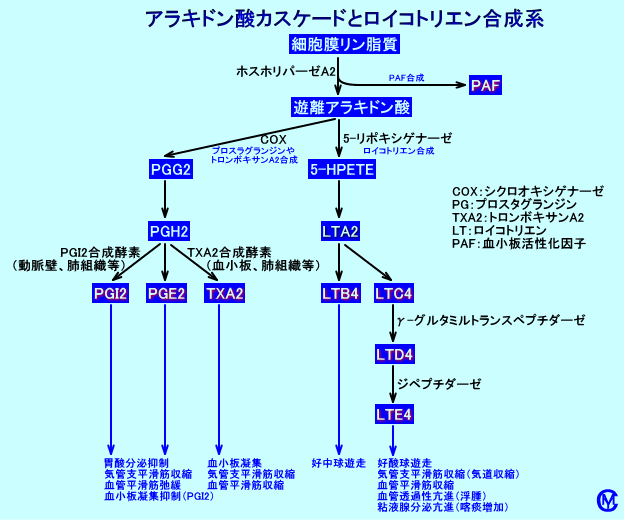
アラキドン酸カスケード
PGE2は、細胞膜のリン脂質に結合しているアラキドン酸から、生成される。
PGE2は、発熱作用や、ブラジキニンによる発痛増強作用(疼痛)や、弱い血管透過性亢進作用(腫脹)や、強い血管拡張作用(発赤、熱感)があり、炎症促進作用を示す。
他方で、PGE2は、マクロファージなどの細胞からサイトカインが産生される際、同じサイトカイン産生細胞から分泌され、サイトカインの産生を抑制し、炎症抑制作用(抗炎症作用、免疫抑制作用)を示す。
アラキドン酸(肉食など、食事に由来する)は、細胞膜のリン脂質(PC)のC2位に、エステル結合している。アラキドン酸は、ホスホリパーゼA2(ホスフォリパーゼA2:PLA2)により、細胞内に遊離される(注1)。
遊離アラキドン酸は、アラキドン酸カスケードと呼ばれる代謝経路でシクロオキシゲナーゼ(COX、cyclooxygenase)により代謝され、PGG2を経て、発痛物質であるPGE2などが合成される。また、LT(ロイコトリエン)合成系で、炎症促進作用(気管支平滑筋や血管透過性亢進作用など)があるLTが合成される。
消炎鎮痛剤(NSAIDsなど)は、PGE2合成を阻害し、痛み(疼痛)と腫脹を抑制する。
PGE2は、生体を発熱させ、組織に腫脹、浮腫などを来たす炎症促進作用がある。反面、PGE2は、TNF-αや産生を抑制し、炎症抑制作用も、ある。
A.シクロオキシゲナーゼ回路
COX(PGH2 Synthase:PGH2シンターゼ)は、アラキドン酸に酸素分子を付加するリポキシゲナーゼであり、PGG2からPGH2を合成するペルオキシダーゼ活性も有する。つまり、単一の酵素、PGエンドペルオキシド合成酵素に、シクロオキシゲナーゼ活性とヒドロペルオキシダーゼ活性がある。
その結果、生理機能の維持や免疫的炎症反応に関与するプロスタグランジン(prostaglandin:PG)や、トロンボキサンA2(thromboxane:TXA2)が血小板や好中球で、合成される。
NSAIDsは、COXの活性を阻害し、アラキドン酸からPGH2が合成されるのを阻害し、プロスタグランジン合成とトロンボキサン合成を抑制する。
副腎皮質ステロイドホルモン(ステロイド剤)は、COXの合成を阻害して、抗炎症作用、鎮痛作用などを現す
なお、 活性酸素の一重項酸素が、PGG2からPGH2が合成される際に産生される。
B.リポキシゲナーゼ経路
アラキドン酸は、5-リポキシゲナーゼにより代謝されて、5-HPETEを経て、免疫的炎症反応に関与するロイコトリエン(leukotrien:LT)が肥満細胞などの白血球で、合成される。
細胞の刺激に応じて、エイコサノイドは、アラキドン酸から合成される。エイコサノイドは、局所で作用した後、速やかに代謝される。
エイコサノイドは、生体の局所で合成されて、局所でホメオスタシス(生体の恒常性)を維持するために働いている。
エイコサノイドは、エイコサペンタエン酸(EPA)からも合成される。
アラキドン酸代謝物(PGやTX)は、生体内では、非酵素的に分解され、半減期が、極めて短い。37℃、pH7.4の水溶液中での半減期は、PGG2やPGH2が約5分、PGI2が約2分、TXA2が約40秒。
1.プロスタグランジン(PG)
PGは、赤血球を除く全ての細胞で産生される。
PG には、AからJまで、10種類、知られている。
PGI2は、PGH2からPGI2合成酵素により生成される。それ以外のPGA2、PGB2、PGC2、PGD2、PGE2、PGF2、PGJ2(JapanのJ:日本で発見された)は、PGH2からPGD,E,F合成酵素により生成されると言う。
ほとんど全ての細胞は、細胞膜にアラキドン酸を含んでいて、プロスタグランジン合成能を有する。
一つの細胞が、全ての種類のPGを産生するのではない。細胞は、その細胞の機能に応じた、1~2種類のPGやTXを、産生する。
NSAIDsやステロイド剤は、COXを阻害し、プロスタグランジン合成を抑制し、抗炎症作用などを現す。
プロスタグランジンは、アポトーシスを抑制する。
表1 化学伝達物質の生理活性の比較
化学伝達物質 TXA2 PGI2 PGE2 SRS-A LTB4 PAF Histamine SP 産生細胞 血小板など 血管内皮細胞など 腎、胃、肺、肝臓など 肥満細胞、好酸球など 肥満細胞など 肥満細胞、単球・マクロファージなど 肥満細胞 C線維 気管支平滑筋 収縮 弛緩 弛緩 収縮 収縮 収縮 収縮 収縮 気道粘液分泌 不明 - 抑制 亢進 無し 亢進 亢進 亢進 胃酸分泌 (抑制) 抑制 抑制 - - - 亢進 - 好中球遊走作用 不明 - - - 有り注2) 有り - 有り 毛細血管透過性 無し 亢進 亢進 亢進 亢進 亢進 亢進 亢進 血管平滑筋 収縮 弛緩 弛緩 収縮 - 収縮 収縮 拡張 血小板凝集 促進 抑制 - - - 促進 - - 1).PGE2
PGE2には、炎症を促進する側面(炎症作用)と、炎症を抑制する側面(抗炎症作用)とが、ある。
a.炎症を促進する側面:発痛させる(疼痛を起す)。血管透過性を亢進させ、腫脹・浮腫などを来たす。発熱させる。
b.炎症を抑制する側面:T細胞からのインターロイキン-2(IL-2)やインターフェロン-γ(IFN-γ)の産生や、TNF-α及びIL‐1の産生や、ロイコトリエン(LT)など他の炎症メディエーターの産生を抑制し、抗炎症作用(免疫抑制作用)を来す。細胞膜を安定化させる。
PGE2は、精のう腺、腎髄質、肺、胃、肝臓などでアラキドン酸より生成される。
主な作用には、血管平滑筋弛緩(血管拡張血圧降下、血流増加)、血管透過性亢進、気管支平滑筋弛緩(気管支拡張)、胃酸分泌抑制や胃粘膜血流増加や胃粘液分泌促進(胃粘膜保護)、血小板凝集抑制、子宮収縮、体温調節(発熱)、細胞膜安定化、覚醒などがある。
消炎鎮痛剤(NSAIDsなど)は、PGE2合成を阻害し、痛み(疼痛)と腫脹を抑制する。
PGE2は、血管透過性を亢進させ、腫脹を起こす:
炎症時に放出されるPGE2は、局所の血流を増加させ、ブラジキニンとともに血管透過性を亢進させ、浮腫形成(腫脹)と炎症細胞の浸潤を増強させる。
PGE2の血管透過性亢進作用は弱いが、ブラジキニンやヒスタミンの血管透過性亢進作用を、増強する。
PGE2は、疼痛を起こす(発痛を増強する):
PGE2は、侵害受容器(ポリモーダル受容器)の感受性を亢進させ、C線維に流れるインパルスを誘発させ、発痛させる。
PGE2の、発痛作用(知覚神経刺激作用)、血管透過性亢進作用、細動脈拡張作用は、弱い。PGE2は、ブラジキニンによる発痛(疼痛)を増強する(発痛増強物質)。
ブラジキニンは、神経線維終末付近(ポリモーダル受容器)で、PGE2を産生させ、PGE2は、EP3受容体を介して、痛みを増強させる。
PGE2、PGD2は、ラットの脳幹部において、高用量投与により痛覚鈍麻作用を、低用量投与により痛覚過敏作用を示す。PGE2、PGD2は、ラットの脊髄において、痛覚過敏作用のみを示し、痛覚鈍麻作用は示さないという。
PGE2は、熱感や発赤を起こす:
PGE2には、PGE1、PGI2と同様に、極めて強い血管拡張作用があり、炎症時に、血管を拡張させ、皮膚の熱感や発赤を起こす。
PGE2は、発熱を起こす:
PGE2は、視床下部の体温調節中枢に作用して、体温のセットポイントを上昇させ、熱産生が増加するので、発熱が起こる(注3)。
気管支喘息の発作は、発熱時に弱まる傾向があるのは、PGE2により、ロイコトリエン産生が抑制される為と考えられる。
PGE2は、抗炎症作用(免疫抑制作用)を有する:
PGE2は、マクロファージなどの細胞からサイトカインが産生される際、同じサイトカイン産生細胞から分泌され、サイトカインの産生を抑制し、抗炎症作用を示す。
PGE2は、T細胞からのインターロイキン-2(IL-2)やインターフェロン-γ(IFN-γ)の産生や、ロイコトリエン(LT)など他の炎症メディエーターの産生を抑制し、抗炎症作用を有する。
炎症時にマクロファージ(単球)から産生される、IL-1やTNF-αは、PGE2の産生を誘導する。
PGE2は、マクロファージからのTNF-αやIL-6の遊離を抑制する(負のフィードバックをかける)。なお、PGE2は、マクロファージからのIL-1の遊離をは、促進する(正のフィードバックをかける)。
(PGE2や、 PGI2は、EP2、EP4受容体を介して、細胞内cAMPを上昇させ、情報伝達系の制御によって、炎症性サイトカインのTNF-α及びIL‐1の産生をは抑制し、また、IL-6やIL-10の産生をは促進する。)
このように、炎症時にマクロファージ(単球)から産生されるPGE2は、抗炎症的に作用する面がある。
マスト細胞(肥満細胞)で産生されたPGE2は、マスト細胞からのヒスタミン遊離(放出)を抑制する。好酸球で産生されるPGE2やPGE1は、ヒスタミンの放出を抑制する。
PGE2は、視床下部に働いて、副腎皮質刺激ホルモン(ACTH)の分泌を増加させ、副腎からのステロイドホルモンの産生を増加させる。
PGE2は、NK細胞活性を抑制する。
このように、PGE2は、炎症の初期に分泌されて、発熱を促す一方で、炎症を解決(the resolution of inflammation)するようにも指令する。
以上をまとめると、PGE2の抗炎症作用は、IL-2、IFN-γ、LT、TNF-α、ヒスタミンの産生や放出を抑制し、ステロイドホルモンの産生を増加させ、NK細胞活性を抑制し、IL-10の産生を促進することによって、生じる。
PGE2には、睡眠阻害作用(覚醒作用)がある。
PGE2は、赤血球変形能を、弱いながら低下させる。
PGE2は、疼痛を起す:PGE2は、PGE1、PGI2と同様に、発痛増強物質(疼痛増強物質)であり、侵害受容器(ポリモーダル受容器)の痛覚閾値を低下させ、痛覚過敏にさせる。ブラジキニンの発痛作用を増強する。
PGI2の発痛増強作用は、15~30分でピークに達し、1時間以内に消失する。それに対して、PGE2の発痛作用は、遅れて出現して、3時間以上続くとされる。
ブラジキニンは、ホスホリパーゼA2を活性化させ、PGE2の産生を刺激する。
抜歯をすると、プロスタグランジンは、抜歯直後から、歯周組織で急速に増加し始め、3~5時間後にピークに達し、7時間後に、元のレベルまで、減少する。抜歯後の痛みは、3~5時間後に、ピークになるが、痛みを感じてから、鎮痛剤を飲んでも、鎮痛剤の効果が得られるまで、速くても15分は要する。抜歯に際しては、抜歯前に、予め鎮痛剤を服用した方が、痛みを遮断するにには、望ましいと言う。
COX-2の主要産物は、PGE2。
PGE2は、細胞内のcAMPレベルを上昇させる。cAMPは、ポジティブフィードバックで、COX-2を誘導する:COX-2誘導→PGE2産生→cAMPの上昇→COX-2誘導
PGE2で上昇したcAMPは、VEGF(vascular endothelial growth factor)やアンジオポエチン2(Ang2)の生成を誘導し、血管新生を増強する。
PGE2は、adenylate cyclaseを賦活させ、cAMPの生成を盛んにする。
cAMPは、血小板内のCa2+濃度を低下させ、actomyosinの収縮を抑制し、血小板凝集を抑制する。
PGE2は、破骨細胞による骨吸収を刺激する。
IL-1は、滑膜細胞に働いて、滑膜細胞の増殖と、PGE2の産生を、増加させる。IL-1やPGE2は、破骨細胞を刺激し、骨吸収を促進させる。関節リウマチなどの疾患で、炎症関節局所の骨萎縮が起こるのは、炎症により、単球・マクロファージが、免疫複合体、補体成分、T細胞から分泌されるIFN-γに刺激され、IL-1やPGE2を産生し、これらのIL-1やPGE2が、破骨細胞を刺激し、骨吸収を促進させる為と言われる。
PGE2は、粘膜上皮細胞内のcAMP濃度を上昇させ、水や電解質(Na+、K+、Cl-)の分泌を亢進させ、下痢を起こす。
PGE2、PGD2、PGF2α、LTC4、LTD4は、腸管平滑筋を強く収縮させるので、下痢を起こす。
PGE2は、腎では、腎血管(輸入細動脈)を拡張させ、腎血流を増大させ、また、近位尿細管でのNa+再吸収を減少させ、利尿させる:PGE2は、Na+と水を排泄させる。
腎臓では、PGE2は、Na+-K+-ATPaseを抑制して、尿細管でのNa再吸収を抑制する:PGE2は、尿細管では、Gαi(抑制性G蛋白(Gαi蛋白)を介して、cAMPの産生を抑制し、PKA(Aキナーゼ:注4)によりNa+-K+-ATPaseが刺激されることを抑制し、Na再吸収を抑制する。(腎臓の糸球体では、高濃度のPGE2は、輸入細動脈を拡張させ、レニン分泌を増加させる(Na再吸収を促進させる)。PGE2は、腎不全では、産生が増加し、腎糸球体の細動脈を拡張させ、糸球体血流量を維持している。)
NSAIDsにより、PGE2の産生が低下すると、腎臓のNa排泄能が低下する(Na再吸収が、抑制されない。その為、浮腫が生じる。NSAIDsにより、腎糸球体血流量が、低下する。)
NSAIDsのインドメタシン(IND)は、糸球体の輸入細動脈では、低濃度のアンジオテンシンII(AII) による動脈収縮作用を増強し、輸出細動脈では、高濃度のAT II による動脈収縮作用を増強したという。これは、NSAIDsが、PGE2の産生を抑制し、PGE2による、輸入細動脈や輸出細動脈の拡張作用を、抑制するためと解釈される。
PGE2は、腎では、以下のような作用を有している。
1.腎の微小血管、特に、輸入細動脈を拡張させて、腎血流や腎糸球体濾過率(GFR)を上昇させる。また、腎髄質の血流を調節する。
2.緻密斑(マクラデンサ)で産生され、レニン分泌を増加させ、血管を収縮させたり、血圧を上昇させる(アルドステロンを介して、Na+と水の再吸収を促進する)。
3.遠位尿細管で、Na+と水の再吸収を抑制する。
4.集合管で、抗利尿ホルモン(バゾプレシン)の作用に拮抗し、水の透過性を抑制し、水の再吸収を抑制する。
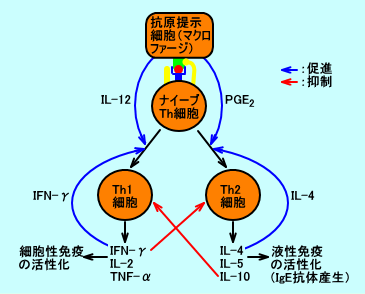
表2 腎臓に於けるPGの作用(参考文献の大野岩男氏の表3を改変し引用) 産生部位 PG 生理作用 細動脈 PGI2、PGE2 腎血流量維持、輸入際動脈拡張 糸球体 PGI2、PGE2、TXA2 腎糸球体濾過率(GFR)調節 遠位尿細管 PGE2、PGI2 バゾプレシン抑制(水の再吸収抑制) PGE2 Na利尿(Na+と水の再吸収抑制) 傍糸球体装置 PGE2、PGI2 レニン分泌増加(Na+と水の再吸収増加) PGE2は、マクロファージが抗原をT細胞に提示する際に分泌されて、ナイーブヘルパーT細胞を、Th2細胞(T helper 2 cell)細胞に分化させる。マクロファージから分泌されるIL-12は、ナイーブヘルパーT細胞を、Th1細胞に分化させる。Th1細胞からは、IFN-γ、IL-2が、Th2細胞からは、IL-4、IL-5が、分泌される。さらに、IFN-γは、Th1細胞の分化を誘導し、IL-4は、Th2細胞の分化や、B細胞のIgEなどの抗体産生を誘導する。
PGE2やPGF2αは、重炭酸イオンの分泌を促進し、胃酸を中和するという。
PGE2の受容体には4種類のEP受容体(EPレセプター:EP1、EP2、EP3、EP4)があり、生体内で異なる臓器や組織に存在して、異なるシグナルを細胞内に流すため、標的組織により異なる作用が発現する。
PGE2は、EP2受容体を介して、cAMP濃度を上昇させ、気管支拡張、血管拡張を来たす。
PGE2は、EP3受容体(EP3受容体)を介して、cAMP濃度を低下させ、平滑筋収縮、発熱、末梢での発痛を来たす。
PGE2は、EP4受容体を介して、cAMPを増加させ、肥満細胞からのヒスタミン遊離の抑制、血小板凝集の抑制が、起こる。PGE2は、EP3受容体を介しては、cAMPを減少させる。
腎臓では、EP1受容体は、集合管に、EP3受容体は、遠位尿細管に、EP4受容体は、腎糸球体と腎動脈に分布している。
PGE2は、腎臓では、EP3受容体(EP3AとEP3B)を介して、cAMPの産生を抑制し、PKAによるNa+/K+-ATPaseの発現を抑制して、尿細管でのNa+再吸収を抑制する。EP3A受容体(EP3Aレセプター)は、遠位尿細管細胞に存在し、バソプレシンのV2受容体(V2レセプター)と拮抗して、cAMPを低下させる。EP3B受容体(EP3Bレセプター)胞は、カルシウムのシグナルトランスダクションを介して、細胞内カルシウムを増加させて、利尿やナトリウム利尿(Na+の排泄)を生じさせるという。
NSAIDsは、PGE2の産生を抑制するので、Na+再吸収が増加し、Naと水分が、貯留して、浮腫などの、副作用を示すことがある。
IL-1βは、NF-kBの発現を介して、COX-2と、EP3受容体のmRNAの発現を、増加させる。
EP3受容体は、肝臓、腎臓、脂肪(epididymal fat)、膵臓のランゲルハンス島では、最も、多く存在するEP受容体。膵臓のランゲルハンス島では、EP1受容体が、EP3受容体に次いで、多く存在する。
骨格筋には、EP3受容体は、少なく、EP2受容体が、多く存在する。
マウスの実験結果では、EP1受容体が欠損すると、ストレス環境で、異常行動を起こすと言う:高所から飛び降りてしまう。
表3 EP受容体 サブタイプ AC 細胞内伝達系 分布臓器(作用) EP1受容体 - Ca濃度上昇 肺、腎集合管 EP2受容体 活性化 cAMP濃度上昇 気管支や血管の平滑筋(弛緩)、上皮細胞(分泌)、肥満細胞(遊離抑制)、知覚神経終末(熱感受性増大) EP3受容体 抑制 cAMP濃度低下 自律神経終末(遊離抑制)、脂肪組織(HSLによる中性脂肪分解抑制)、胃(胃酸分泌を弱く抑制)、胃腸管や子宮や血管の平滑筋(収縮)、腎遠位尿細管(Na+再吸収抑制)、発熱 EP4受容体 活性化 cAMP濃度上昇 静脈、気管、子宮、動脈管(弛緩)、免疫抑制 PGE2やIL-1βは、グルコース(ブドウ糖)によるインスリン分泌を、抑制する。
COX-2により産生されたPGE2は、EP3受容体を介して、アデニル酸シクラーゼ(AC)を抑制するので、cAMPが減少し、インスリン分泌が、抑制される。
IL-1βは、NF-kBの発現を介して、COX-2を発現させたり(PGE2の産生が増加する)、EP3受容体のmRNAを発現させるので、インスリン分泌が、抑制される。
サリチル酸は、IL-1βによる、NF-kBの発現を抑制し、COX-2や、EP3受容体(EP3受容体)のmRNAの発現を妨げて、PGE2によるインスリン分泌抑制作用を抑制するので、インスリン分泌が、増加し、低血糖になることがある。
なお、アスピリンは、IL-1βによる、NF-kBの発現を抑制しないので、インスリン分泌に影響するのは、アスピリンでなく、サリチル酸と考えられる。
PGE2は、肝臓で、アデニル酸シクラーゼ(AC)を抑制し、グルカゴンによるグリコーゲン分解(glycogenolysis)を、抑制する。.
PGE2には、動脈管拡張作用がある。NSAIDs(Indomethacin)は、未熟児に投与すると、PGE2を抑制して、動脈管を閉鎖させる。
卵胞のCOX-2で合成されるPGE2は、排卵を促進させるので、NSAIDsの長期間服用は、不妊の原因になる。
PGE2は、妊娠子宮を収縮させるが、非妊娠子宮を弛緩させると言う。
ビタミンEは、PGE2の合成を抑制する。
アスピリン喘息と言って、喘息患者がアスピリンを内服すると、喘息発作が誘発されることがある。この機序として、 気管支に分布するPGE2(気管支を拡張し、細胞膜安定化作用がある)の産生が、アスピリンにより抑制され、気管支が収縮したり、肥満細胞からヒスタミンなどが遊離されることが、考えられる。また、アスピリンは、PG合成系を阻害するが、リボオキシゲナーゼ経路には影響しないので、強力な気管支収縮作用のあるロイコトリエン(LTC4、LTD4、LTE4)の産生が増加して、アスピリン喘息が起こることも、考えられる。
PGE2は、脂肪組織では、EP3受容体を介して、アデニル酸シクラーゼ(AC)を抑制し、cAMPの産生を抑制し、ホルモン感受性リパーゼ(HSL)による、中性脂肪の分解を、抑制する(肥満になる)。
サリチル酸は、COX-2によるPGE2の産生や、EP3受容体発現を抑制して、PGE2によるインスリン分泌抑制作用を抑制するので、インスリン分泌が、増加し、低血糖や肥満を招くおそれがある(サリチル酸は、PGE2産生抑制によるHSL抑制解除作用より、インスリン分泌促進によるHSL抑制作用を示す)。
なお、アスピリン系の薬(サリチル酸を含む)は、プロスタグランジン(PGE2など)の産生を阻害し、交感神経緊張にして、代謝を亢進させ、運動をしなくても、エネルギーが消費されるので、痩せるとする人もいる。
女性ホルモン(エストロゲン)のエストラジオール(E2:活性化エストロゲン)は、子宮内膜症組織で、COX-2の発現を誘導し、PGE2の産生を、増強させる。PGE2は、アロマターゼの発現を増強させ、アンドロステンジオン(A)を、エストロン(E1)への変換を促進させる。エストロン(E1)は、17β-hydroxysteroid dehydrogenase type 1により、エストラジオール(E2)に変換される。
このように、子宮内膜症組織では、女性ホルモン(エストロゲン)により産生が促進されたPGE2は、アロマターゼの発現を増強させ、女性ホルモン(エストロゲン)の産生を増加させる(positive feedback loop)。
NSAIDsのCOX-2阻害薬は、子宮内膜症病巣を縮小させたり、VEGF産生を低下させる作用がある(NSAIDsは、子宮内膜症の疼痛治療に有用)。
子宮内膜症病巣部には、肥満細胞が浸潤し、脱顆粒により、ロイコトリエン(LT)が放出され、線維芽細胞が産生され、癒着が起こる。抗ロイコトリエン受容体拮抗薬(LTRA)は、疼痛の原因となっている器質性病変(腹腔内癒着など)を予防するのに、有用と言われる(抗ロイコトリエン受容体拮抗薬は、子宮内膜症の疼痛予防に有用)。
プロスタグランジン(PGE2)は、気管支や血管の平滑筋の弛緩作用、脂肪組織でのHSLによる中性脂肪分解抑制作用などがある。
プロスタグランジン(PGE2)は、交感神経を抑制し、カテコールアミン(アドレナリンなど)の産生を抑制する。
プロスタグランジン(PGE2)は、無水エタノール、強塩酸、強アルカリ、高浸透圧溶液、熱傷などによる壊死を起こす物質や要因から、胃粘膜傷害を阻止する(Robert等の提唱したサイトプロテクションの概念)。
胃粘膜病変を起こす作用は、病変の広がりで判断すると、水浸拘束ストレス<酸性アスピリン<無水エタノールの順に強い。また、胃粘膜病変を起こす作用は、病変の数で判断すると、水浸拘束ストレス≧酸性アスピリン>>の順に弱い:酸性アスピリンは、胃粘膜病変を数多く起こすが、病変の広がり(大きさ)は、無水エタノールで起こる胃粘膜病変より、狭い。水浸拘束ストレスは、胃粘膜病変を数多く起こすが、病変の広がり(面積)は、狭い。無水エタノールは、広い胃粘膜病変を起こすが、数は少ない。
PGE2は、脂肪組織では、EP3受容体を介して、アデニル酸シクラーゼ(AC)を抑制し、cAMP濃度を低下させ、HSLによる中性脂肪分解を抑制する(インスリンと同様の作用)。
PGE2は、肝臓では、アデニル酸シクラーゼ(AC)を抑制し、グルカゴンによるグリコーゲン分解を、抑制する(インスリンと同様の作用)。
PGE2は、膵臓では、EP3受容体を介して、グルコースによるインスリン分泌を、抑制する。
PGE2は、腎臓(集合管)では、cAMP濃度を減少させ、バゾプレシン(vasopressin)による水の再吸収を抑制する。
PGE2は、骨格筋では、多量に、EP1受容体や、EP2受容体が存在し、筋線維の収縮と弛緩の両方に、関与する。
PGE2は(アデニル酸シクラーゼを抑制し、細胞内cAMP濃度を低下させる)は、低濃度で、気管支平滑筋を弛緩させる。
PGE2は、迷走神経末端からのアセチルコリン(グアニル酸シクラーゼを活性化させ、細胞内cGMP濃度を上昇させる)の遊離を抑制し、気道反応性を減弱させる(アセチルコリンによる気管支平滑筋の収縮を抑制する)。
肥満細胞では、エピネフリン(交感神経)によってアデニル酸シクラーゼが活性化され、細胞内cAMP濃度が上昇すると、ヒスタミン遊離が抑制され、アセチルコリン(副交感神経)によって、グアニル酸シクラーゼが活性化され、細胞内cGMP濃度が上昇すると、ヒスタミン遊離が促進される。
熱傷モルモットに、熱傷直後から、14日間、リノール酸(n-6系の多価不飽和脂肪酸)を投与すると、PGE2の産生が、高まった。
PGE2により、細胞性免疫(Th1細胞)が抑制される。
ラット・カレニゲン胸膜炎で、胸水中のプロスタグランジン等を測定すると、まず、6-keto-PGF1α(PGI2の分解産物)が、カラニゲン溶液注射1時間後をピークに、胸水中に増加し、その後、急激に減少する。次に、PGE2が注射3時間後をピークに、胸水中に増加し、次第に減少する(9時間後には、かなり低値になる)。さらに、TXB2(TXA2の分解産物)が、注射3~7時間後をプラトーに、胸水中に増加する:PGI2→PGE2→TXA2の順に生成される。
ラット・カレニゲン胸膜炎では、PGE2やブラジキニン(BK)により血管透過性が亢進し、特に、注射1~3時間後まで、胸水が増加する。
PGE1やPGE2の血管透過性亢進作用は弱いが、ブラジキニン(BK)の血管透過性亢進作用を増強させる。閾値量のPGE2(0.1μg)を、BK(0.1μg)と共に皮内注射すると、BKの血管透過性亢進作用が、十倍増強される。ヒスタミンは、BKの血管透過性亢進作用を増強させない。
多くの免疫反応は、cAMPにより、抑制される。
白血球のライソソーム酵素などの放出は、PGE1(やPGE2)など、cAMPを上昇させる因子により抑制され、cGMPを上昇させる因子により促進される。
好中球の遊走能(走化性)は、cAMPを上昇させる因子(PGE2など)により抑制され、cGMPを上昇させる因子により促進される。しかし、マクロファージ(単球:大食細胞)の遊走能(走化性)は、反対に、cAMPを上昇させる因子(PGE2など)により促進され、cGMPを上昇させる因子により抑制される。
PGI2やPGE2は、cAMPを介して、ライソゾーム酵素の分泌(遊離)を抑制する(antiinflammatory effect:抗炎症作用)。
PGE1やPGE2は、好中球内cAMP濃度を上昇させ、好中球からのライソゾーム酵素(エラスターゼなど)の遊離を、抑制する(ライソゾーム膜安定化作用)。
PGE2は、コラーゲン合成を促進させる。
2).PGI2(プロスタサイクリン)
PGI2(prostacyclin)は、血管内皮細胞から産生され、血小板凝集を抑制したり、血管平滑筋を弛緩させて血管を拡張させたりして、血管内の血流の維持に関与する。
血管内皮細胞から産生されるPGI2は、血小板凝集や血管平滑筋に対して、血小板から産生されるTXA2とは相反する作用を示し、恒常状態では、この両者の生成のバランスが保たれている。
PGI2には、肺でも生成され、気管支拡張作用(気管支平滑筋を弛緩させる)、肺血管拡張作用もある:一般にPGは、肺の細胞内で酸化され(-OHが=Oになる)、生理活性を失う。しかし、PGI2は、肺の細胞内に入りにくいので、肺ではほとんど代謝されない。
PGI2は、胃でも産生されて、胃粘膜の血流を維持したり、粘液産生を増加させ、胃粘膜を保護している。
PGI2は、腎でも生成される。
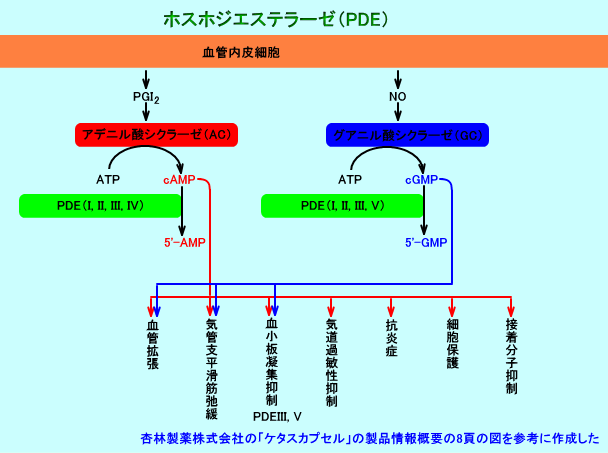
PGI2は、アデニル酸シクラーゼ(AC:アデニルサイクラーゼ)の活性を亢進させる。アデニル酸シクラーゼは、ATPからcAMPを生成する。cAMPは、血小板内のCa2+濃度を低下させ、血小板の収縮や放出反応を抑制する。cAMPは、血小板膜のリン脂質からアラキドン酸が遊離されるのを抑制し、また、アラキドン酸からのトロンボキサンA2(TXA2)の生成を抑制する。このようにして、PGI2は、cAMPを生成させ、血小板内のCa2+濃度を低下させ、血小板凝集を抑制する。
PGI2には、血小板凝集塊を解離させる作用もある。
PGI2は、cAMP濃度を上昇させ、強力な血小板凝集抑制作用を現す。
血小板凝集抑制作用は、PGI2>PGD2>PGE2の順に、弱い。
PGI2は、発痛増強物質である。
PGI2は、PGE2より、ブラジキニン(BK)による発痛(疼痛)を増強する作用が、強い。
PGI2は、細胞のcAMP濃度を増加させ、抗炎症的に作用する。
PGI2は、PGH2からprostacyclin synthetaseにより、生成される。
PGI2は、不安定で半減期が短く(37℃、pH7.4の水溶液中での半減期は、約2分)、不活性な6-keto-PGF1αにすぐ分解される。
PGI2は、コレステロールを分解する酵素、ACEH活性を促進する。
PGI2の受容体は、IP受容体とのこと。
経口プロスタサイクリン誘導体製剤(ベラプロストナトリウム:医薬品名ドルナー)には、血管内皮細胞保護作用があり、過酸化脂質による血管内皮細胞障害を抑制する。
深呼吸(静座など)をすると、肺の肺胞が膨らんで、肺胞の毛細血管からPGI2が放出され、血液中のPGI2濃度が、上昇する。
PGI2やPGE2は、cAMPを介して、ライソゾーム酵素の分泌(遊離)を抑制する(antiinflammatory effect:抗炎症作用)。
3).PGE1
PGE1には、動脈管拡張作用や血小板凝集抑制作用がある。(注射で新生児期の動脈管開存症に用いた時の副作用には、発熱、下痢、出血傾向、などがある。)
PGE1は、胃酸分泌を抑制し、発熱させるという。
60分間膵臓を阻血する実験で、阻血する前にPGE1を30分間持続静脈注射すると、膵臓細胞は保護され、インスリン分泌能や外分泌能(アミラーゼなど)の傷害は、軽減される(細胞膜安定化作用)。
肥満細胞や、好塩基球は、細胞内のcAMP濃度が減少すると、微弱な刺激によっても、ヒスタミン遊離が起こる(アトピー性皮膚炎の痒みが出現し、易刺激性が増強する)。
プロスタグランジン(PGE1など)や、アドレナリン(エピネフリン)は、アデニル酸シクラーゼを活性化させ、細胞内のcAMP濃度を上昇させるので、ヒスタミン遊離が起こりにくくなる(細胞膜安定化作用)。
PGE1は、肺や全身の血流を増加させ、肺障害などの臓器傷害を防止する。
ラットを用いた実験で、リポ多糖類(LPS:200μ/kg)を、腹腔内に投与し、4時間後に、PGE1(5μ/kg/分で1分間)を、頚静脈から注射し、同時に、標識した好中球を注射し、2分後の臓器の状況を、顕微鏡で観察した。
その結果、LPSを投与した後、PGE1を投与(注射)しないと、肺や肝臓で、好中球が、接着していた。
しかし、PGE1を投与(注射)した場合、肺や肝臓で、接着している好中球が、減少した。
イヌを用いた実験で、サイトカイン(IL-1やTNF-α)を、持続的に静脈内注射すると、末梢血中の白血球数が減少し、臓器に集積する。集積(接着)する好中球の数は、肺が圧倒的に多く、肝臓や、腎臓に接着する好中球数は、肺より少ない。
PGE1は、抗体産生を亢進させる。
PGE1やPGE2は、好中球内cAMP濃度を上昇させ、好中球からのライソゾーム酵素(エラスターゼなど)の遊離を、抑制する(ライソゾーム膜安定化作用)。
PGE1は、ラットの免疫複合体による血管炎を抑制する。
PGE1は、好中球の遊走(chemotaxis)を抑制し、好中球による免疫複合体の貪食や、ライソゾーム酵素の放出(遊離)を抑制する。その結果、血管内や血管周囲に、免疫複合体や補体は沈着するが、好中球からライソゾーム酵素が遊離されないので、ライソゾーム酵素による組織障害が、起こらない。
4).PGF2α
PGF2αは、マクロファージ、白血球で、生成される。
PGF2αには、子宮収縮作用(生理痛の原因となる)、消化管粘液分泌促進、腸管蠕動亢進作用、気管支平滑筋収縮作用、粘液分泌亢進作用がある。
なお、子宮宮筋層は主としてPGI2、TXA2を産生し、子宮内膜は主として PGE2とPGF2αを産生するという。
PGF系のプロスタグランジンは、細胞内cGMP(cyclic GMP)濃度を上昇させる。
5).PGD2
PGD2は、ヒト肺実質、肥満細胞で産生される。
PGD2には、血管平滑筋収縮作用、血管透過性亢進作用、気管支平滑筋収縮作用、粘液分泌亢進作用があるが、血小板凝集は抑制する。
PGD2には、自然睡眠誘発作用がある。カゼなどで発熱した時に、眠くなって、体を休息させようとする。
PGD2は、アレルギー性鼻炎で、ヒスタミン、LT、PAFと同様に、鼻粘膜の血管に直接作用して、血漿を漏出させ、鼻汁成分にさせる。
補体C5aの食欲増進作用(摂食促進作用)には、PGD2が関与している。
PGD2は、PGH2より、PGD合成酵素により合成される。
PGD2の代謝産物の15-deoxy-PGJ2は、脂肪細胞の分化や、肥満に関与すると言う。
6).PGG2、PGH2
PGG2、PGH2には、血小板凝集促進作用や、血管平滑筋収縮作用、気管支平滑筋収縮作用がある。
2.トロンボキサンA2(TXA2)
TXA2は、血小板で、トロンボキサン変換酵素により生成される。
TXA2には、血小板凝集作用、血管平滑筋収縮作用、気道平滑筋収縮作用がある。
生体は、外傷などで血管が損傷を受けた時に、血小板から、TXA2を放出させ、血小板凝集や血管収縮を起させ、止血しようとする。
TXA2は、血小板から濃染顆粒を放出させる。
トロンボキサン合成酵素の活性の高い組織は、まず、血小板、続いてマクロファージ、肺、肝臓である。その他の組織にも、低値ながらトロンボキサン合成酵素は、存在する。
アラキドン酸からは、血小板では、トロンボキサン合成酵素により、血小板凝集促進作用があるTXA2が生成される。
同じアラキドン酸から、血管内皮細胞では、血小板凝集阻止作用があるプロスタグランジンI2(PGI2:プロスタサイクリン)が生成される(同じ材料から、同じ代謝経路で、反対の作用を示す生理活性物質が産生されることは、生命の恒常性を保つために、好都合な仕組みと、思われる。)
n-3系の不飽和脂肪酸のEPAが、血小板で代謝されて生成されるトロンボキサンA3(TXA3)は、アラキドン酸が代謝されて生成されるTXA2と異なり、血小板凝集作用や、血管平滑筋収縮作用が無いとされている。
実際、EPAを含む魚食により、血小板凝集能が抑制される。
EPAからは、COX-1により、PGI3も生成されるが、PGI3には血小板凝集抑制作用がある。
魚油(EPAやDHAを含む)を、成人の喘息患者に投与すると、臨床症状は改善しないが、多核白血球のEPAは上昇し、アラキドン酸は減少し、LTB4、C5aは減少すると言う。
TXA2は、血小板内のCa2+濃度を増加させ、血小板を収縮させたり、ADPやセロトニンを放出させ、血小板を凝集させる。また、血小板内のCa2+濃度の増加により、PLA2が活性化され、アラキドン酸からのTXA2の合成が促進される。
TXA2受容体は、PGH2受容体と同じ。
TXA2は、血小板で、膜のアラキドン酸から、PGH2を経て、thromboxane synthetaseにより、生成される。
TXA2は、血中半減期が約30秒(約40秒)と短く、TXB2に代謝される。
慢性糸球体腎炎腎不全群、紫斑病性腎炎活動期では、血漿TXB2は、高値を示し、PGI2の代謝産物の血漿6-keto-PGF1αは、低値を示す。
TXA2は、細胞外Ca2+流入を促進させ、ホスファチジルイノシトール(PI)の分解、ジアシルグリセロール(DAG)の産生を高める。ジアシルグリセロール(DAG)からも、アラキドン酸が遊離される。
表4 エイコサノイドやPAFの産生細胞・組織 産生細胞・組織 COX 5-lipoxygenase PAF PGD2 PGE2 PGF2α PGI2 TXB2 LTB4 LTC4 肺マスト細胞 + - - - - - + + 好塩基球 - - - - - - + + 好酸球 - + - - - - + + 好中球 - + - - + + - + 肺胞MΦ - + - - + + + + 単球 - - - - - + + + 血小板 - - - + + - - + 肺組織 - + + (+) - - - + 気道上皮 + - - - - - + +
3.ロイコトリエン(LT)
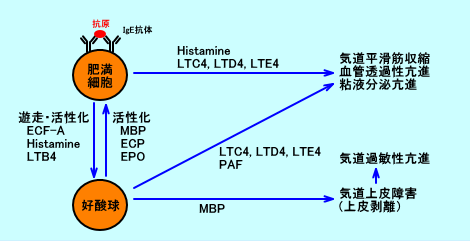
LTC4、LTD4、LTE4は、肥満細胞、好酸球、好中球、単球・マクロファージなどから産生される。
LTC4、LTD4、LTE4は、気管支、血管、消化管の平滑筋を、ゆっくりと、持続的に収縮させる。
LTC4、LTD4、LTE4の気管支平滑筋収縮作用は、ヒスタミンより強力(LTC4は1000倍)に作用し、しかも、その収縮作用は持続する。
また、LTC4、LTD4、LTE4は、気管支喘息の気道炎症に関与する:毛細血管の血管透過性を亢進させ組織内に浮腫を誘導させたり、気道粘膜の繊毛運動を減少させたり、気道粘液の分泌を亢進させたり、好酸球を気道に遊走・活性化させる。
ロイコトリエン合成に関与する5-リポキシゲナーゼは、白血球系細胞(肥満細胞、好塩基球、好酸球、好中球、単球など)に局在している。
LTC4、LTD4、LTE4(CysLT)は、白血球系細胞でも、特に、肥満細胞や好酸球から産生されて、気管支喘息などアレルギー性炎症を引き起こす。LTC4やLTB4は、細胞内で合成された後、能動的に細胞外に輸送され、細胞外に存在するγ-グルタミルトランスペプチダーゼによりLTD4に変換され、更に、ジペプチダーゼによりLTE4に変換される。
LTC4、LTD4、LTE4(CysLT)は、気管支、血管、消化管などの平滑筋収縮作用がある。LTC4、LTD4、LTE4(CysLT)を吸入させると、CysLT1レセプターに結合し、(摘出された)気管支は、数分かけて緩徐に収縮し、数十分間収縮が持続する。CysLTによる気管支収縮の強さは、ヒスタミンやメサコリンによる収縮より10,000倍強い。
LTC4、LTD4、LTE4(CysLT)は、気管支平滑筋収縮作用(気道収縮作用)に加え、血管透過性亢進作用(組織内浮腫誘導作用)、粘液腺分泌亢進作用、平滑筋増殖刺激作用、好酸球遊走作用などを有している(気道過敏性の形成に関与する)。
LTB4には、強力な白血球遊走能亢進作用、及び白血球活性化作用がある。
ロイコトリエン(LT)のLTC4、LTD4、LTE4は、気道(気管支)の炎症細胞(肥満細胞など)から放出され、気管支平滑筋を収縮させる。
ロイコトリエン(LT)のLTC4、LTD4、LTE4は、気管支喘息での気道狭窄(即時型と遅発型)、気道炎症を引き起こす。ロイコトリエン(LT)の合成阻害薬(遊離抑制剤)や、拮抗薬は、気管支拡張作用や、気道炎症抑制作用を現す。
気管支喘息では、抗原(アレルゲン)刺激により、即時型気道収縮(抗原刺激直後から収縮し、15~20分後に最大になり、1時間以内にほとんど消失する)と、遅発型気道収縮(数時間後に起こる)との二相性反応(収縮)を起こす。即時型反応では、肥満細胞から脱顆粒が起こり、ヒスタミンが放出されたり、ロイコトリエン(LT)が新生され、気管支平滑筋が収縮する(気道収縮が起こる)。遅発型反応では、気道局所に遊走・浸潤して来る炎症性細胞(特に好酸球)からロイコトリエン(LT)やPAFなどが放出され、気管支平滑筋が収縮する。ロイコトリエン拮抗薬(LTレセプター拮抗薬)は、ロイコトリエンによる気管支収縮を抑制し、気管支拡張作用を現す。
表5 長期管理薬の特徴(参考文献の吉原重美氏の表を改変し引用) 作用 経口薬 吸入薬 ロイコトリエン
受容体拮抗薬テオフィリン Th2サイトカイン
阻害薬吸入ステロイド薬 DSCG 長期間作用性
β2刺激薬抗炎症作用 ++ + + +++ + - 気管支拡張作用 ++ ++ - - - +++ 抗リモデリング作用 ++ - + ++ - - 末梢気道への作用 ++ ++ ++ + + + 鼻炎合併 +++ - ++ - - - ウイルス性喘鳴 +++ + - - +++ + 慢性咳嗽 ++ + ++ + + - 乳児反復性喘鳴 +++ + ++ ++ +++ - 安全性 +++ + +++ + +++ +
肉や卵に多く含まれるアラキドン酸(注5)や、体内でアラキドン酸に代謝されるリノール酸を過剰に摂取すると、生理活性物質である、TXA2、PG、LTの合成が亢進して、炎症反応の強度が影響を受ける可能性がある。
ホスホリパーゼ阻害剤には、塩酸ジラゼフ (医薬品名:コメリアン )がある。
トロンボキサンA2合成阻害剤には、塩酸オザグレル(医薬品名:ドメナン、ベガ)がある。
ロイコトリエン(LT)の内、分子内にシステインを含むLTC4、LTD4、LTE4は、システイニルLT(CysLT)と呼ばれている。
CysLTは、ヒスタミンの約1,000倍の強力な持続性平滑筋収縮作用を有する。
CysLTは、血管透過性亢進(浮腫)、粘液分泌促進、好酸球浸潤を引き起こし、気道過敏性を亢進させ、喘息の発症、気道炎症に関与する。ウイルス感染(RSウイルスなど)で喘鳴を来たした患児は、鼻汁中のLTC4(CysLT2受容体に結合する)濃度が、有意に上昇していると言う。
CysLT受容体には、LTD4との親和性が高いCysLT1受容体と、LTC4との親和性が高いCysLT2受容体とが存在する。
CysLT1受容体(LTD4が結合する)は、気管支平滑筋や、好酸球など炎症細胞に、多く存在する。LT受容体拮抗薬のモンテルカスト(医薬品名:キプレス、シングレア)は、CysLT1受容体に選択的に結合して阻害する。
アスピリンは、通常の用量や低用量(アスピリン喘息発作を誘発する程度の用量)では、LTC4の産生を増加させる。低用量(低濃度)のアスピリンは、LTC4産生に必要な細胞質型ホスホリパーゼA2(cPLA2:細胞膜からのアラキドン酸遊離を調節する)の活性化を増強させる(LTC4産生も増加する)。
アスピリンは、高用量(細胞毒性を示さない程度の用量)では、LTC4の産生を減少させる。
4.PAF
PAFは、platelet activating factor(血小板活性化因子)の略。
PAFの構造は、1-O-アルキル-2-アセチル-sn-グリセロ-3-ホスホコリン。
PAFには、アルキル基の鎖長が異なる、PAF16:0とPAF18:0の2種類が存在する。
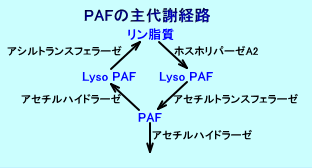
細胞膜のリン脂質が、ホスホリパーゼA2(PLA2)により加水分解され、アラキドン酸が遊離する際に、細胞膜のリン脂質に含まれる1-O-アルキル-2-アシル-GPC(GPC:glycerophosphocholine:グリセロホスホコリン)も、ホスホリパーゼA2(PLA2)により分解され、リゾPAF(1-O-アルキル-GPC)が生成され、さらにアセチルトランスフェラーゼにより、PAFが、生成される。
PAFは、1-O-アルキル-2-アセチル-グリセロールに、CDP(シチジン三リン酸)-コリンが作用しても、生成される。、
と共に、lysophosphoryl cholineが産生され、PAF(platelet activating factor:血小板活性化因子)が合成される。
PAFは、好塩基球、肥満細胞、単球・マクロファージ、好中球、好酸球、血管内皮細胞、血小板など、広範な細胞から放出される。
PAFは、血小板凝集作用、血管透過性亢進作用(ヒスタミンより強力)、平滑筋収縮作用(気管支収縮、回腸収縮)、白血球浸潤作用、好酸球の遊走作用、血圧降下作用がある。
PAFは、血漿中では、速やかに分解され、半減期は、約30秒。
グリセロール骨格の1位がアシル型のPAFは、100分の1以下の活性を有しない。
2位がアセチル基でないPAF様リン脂質(鎖長2~4)は、PAF受容体に結合する。PAF様リン脂質は、酸化LDL中に含まれており、動脈硬化症の発症に関連すると考えられている。
PAF受容体は、プロモーターによりマクロファージや好中球に発現するが、多くの臓器に普遍的に発現している。
注1:ホスホリパーゼA2(ホスフォリパーゼA2、phospholipase A2:PLA2)の加水分解に際し、リゾリン脂質が生成されると、考えられる。
抗アレルギー剤のペミロラストカリウム(pemirolast pottasium:TBX:医薬品名:アレギサール)は、肥満細胞(マスト細胞)に作用し、抗原刺激された際に、G蛋白質の活性化によるホスホリパーゼC(PLC)の活性化を抑制し、イノシトールリン脂質代謝を阻害する。
ペミロラストカリウムは、ホスファチジルコリン(PC)のPLA2による分解を抑制し、アラキドン酸遊離を抑制し(プロスタグランジンやロイコトリエンの産生が抑制される)、1,2-ジアシルグリセロール(DAG)、ホスファチジン酸の産生を抑制する)。
ペミロラストカリウムは、肥満細胞(マスト細胞)内への細胞外Ca2+流入や、細胞内Ca2+の遊離を抑制し、肥満細胞かのケミカルメディエーター遊離(ヒスタミン、ECF-A、NCFなど)を抑制する。
ペミロラストカリウムは、肺組織、鼻粘膜、末梢白血球、腹腔浸出細胞などからの、ヒスタミン、LTB4、LTC4、LTD4、PGD2、TXA2、TXB2、PAFなどの遊離を抑制する。ペミロラストカリウムは、肺組織(肺切片)からの6-keto-PGF1α遊離をは、抑制しない。
ペミロラストカリウム(TBX)は、10-6~10-4Mの濃度で、用量依存性に、好酸球の遊走を有意に抑制する。
ペミロラストカリウム(TBX)は、ヒツジ性嚢腺ミクロゾームでのアラキドン酸からのPGE2合成や、モルモット腹腔浸出細胞でのアラキドン酸からの5-HETE生成(5-HPETEから生成される)には、明らかな影響を及ぼさない。
インドタサシンは、1×10-5Mの濃度で、肺組織(肺切片)からのPGD2、TBX2、6-keto-PGF1α遊離を、抑制する。
抗アレルギー剤の塩酸アゼラスチン(医薬品名:アゼプチン)は、細胞外Ca2+流入を抑制し、5-リポキシゲナーゼを阻害し、細胞内cAMPを上昇させ、細胞膜安定化作用を示す。
塩酸アゼラスチンは、モルモットの肺切片、ヒトの好中球や好酸球からのLTC4、LTD4、LTB4の産生・遊離を抑制する(ロイコトリエン産生・遊離抑制作用)。
塩酸アゼラスチンは、ヒトやウサギの好塩基球、ラットの肥満細胞からのヒスタミンの遊離を抑制し、抗ヒスタミン作用を示す(ヒスタミンによる、気管筋、回腸の収縮を抑制する:肥満細胞や好塩基球からのヒスタミン遊離抑制作用)。塩酸アゼラスチンは、LTB4によるヒトの好中球の遊走、PAFによるモルモットの好酸球の遊走・浸潤を抑制する(炎症細胞の遊走・浸潤抑制作用)。また、塩酸アゼラスチンは、モルモットの好中球からの活性酸素の産生を、顕著に抑制する(活性酸素産生抑制作用)。
| 商品名 | アゼプチン | アレギサール | アレジオン | アレロック | オノン | クラリチン | ジルテック | シングレア | ゼスラン | セルテクト | リザベン |
| 一般名 | 塩酸アゼラスチン | ペミロラストカリウム | 塩酸エピナスチン | 塩酸オロパタジン | プランルカスト水和物 | ロラタジン | 塩酸セチリジン | モンテルカスト | メキタジン | オキサトミド | トラニラスト |
| 1日投与量 | 2~4mg | 0.4mg/kg | 10~20mg | 10mg | 450mga)(4cap) | 10mg | 10mg | 10mg | 12mg | 60mg | 300mgc) |
| 1日投与回数 | 2 | 2 | 1 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 3 |
| 投与時間 | 朝食後、就寝前 | 朝食後、就寝前 | 朝、就寝前 | 朝食後、夕食後 | 食後 | 就寝前 | 就寝前 | 朝、就寝前 | |||
| H1受容体拮抗作用 | + | + | + | - | + | + | - | + | + | ||
| LTC4拮抗作用 | + | + | + | ±b) | + | + | |||||
| LTB4拮抗作用 | + | ± | |||||||||
| PAF拮抗作用 | + | + | + | + | |||||||
| セロトニン拮抗作用 | + | - | - | + | + | ||||||
| ブラジキニン拮抗作用 | + | + | + | ||||||||
| ヒスタミン遊離抑制作用 | + | + | + | + | |||||||
| SRS‐A遊離抑制作用 | + | + | + | + | + | + | + | + | |||
| LTB4遊離抑制作用 | + | + | + | ||||||||
| TXB2遊離抑制作用 | + | +(トロンボキサン) | |||||||||
| PAF遊離抑制作用 | + | + | |||||||||
| PGD2遊離抑制作用 | + | + | |||||||||
| ECP・EPX遊離抑制作用 | + | ||||||||||
| タキキニン遊離抑制作用 | + | ||||||||||
| 気管支喘息 | ○ | ○ | ○(DSは×) | - | ○ | - | - | ○ | ○ | - | ○ |
| アレルギー性鼻炎 | ○ | ○ | ○ | ○ | ○ | ○ | ○ | - | ○ | ○ | ○ |
| 蕁麻疹 | ○ | - | ○ | ○ | - | ○ | ○ | - | ○ | ○ | -c) |
a):オノン(プランルカスト水和物)の1日投与量450mgは、4カプセル(1回2カプセルを朝食後及び夕食後の2回内服する)。
注2:LTB4は、好酸球遊走作用もある。
オノンドライシロップ10%は、通常、小児には、ドライシロップとして70mg/kg/日(プランルカスト水和物として7mg/kg/日)を、朝食後及び夕食後の2回に分け、用時懸濁して内服する。オノンドライシロップ10%の1日最高用量は、ドライシロップとして100mg/kg/日(プランルカスト水和物として10mg/kg/日)とし、プランルカスト水和物の成人の通常用量である450mg/日(ドライシロップとして4.5g/日)を超えないこと(オノンドライシロップ10%の1日最高用量は、4.5g/日)。
b):シングレア(モンテルカストナトリウム)は、LTD4拮抗作用は+(LTD4の受容体結合を強力に阻害する)。シングレアは、システイニルロイコトリエン(CysLT1)受容体へのロイコトリエンD4(LTD4)の結合を抑制し、気管支のLTD4による収縮を競合的に阻害する。LTD4は、ヒスタミンより1,000倍強い気道平滑筋収縮作用があり、収縮が持続する。シングレアは、ヒスタミン、アラキドン酸、セロトニン、アセチルコリンによって誘発される気管支収縮を阻害(抑制)しない。シングレアは、小児用にシングレアチュアブル錠5が販売されている。6歳以上の小児は、シングレアチュアブル錠5を1日1回就寝前に内服する。シングレアのようなロイコトリエン拮抗剤(LT拮抗薬)を使用時に、Churg-Strauss症候群様の血管炎(末梢血の好酸球増加,好酸球浸潤をともなう壊死性血管炎や肉芽腫)を生じることがあるの、末梢血,好酸球数、しびれ、四肢脱力、発熱、関節痛、肺の浸潤陰影などの血管炎症状が現れていないか注意が必要。
c):リザベンドライシロップは、通常、小児には、0.1g/kg/日(トラニラストとして5mg/kg/日)を3回に分け、用時懸濁して内服する。リザベン細粒は、通常、小児には、0.05g/kg/日(トラニラストとして5mg/kg)を3回に分け、経口投与する。
リザベン(トラニラスト製剤)は、アトピー性皮膚炎、ケロイド・肥厚性瘢痕に保険適応が承認されている。
注3:感染症に際して、マクロファージなどから産生されるIL-1βは、脳視床下部に作用して、PGE2の産生を亢進させる。産生されたPGE2は、脳視床下部の体温調節中枢に作用して、体温のセットポイントを上昇させ、熱産生を増加させ、生物を発熱させる。
発熱は、ヒトでは、体温を上昇させることにより、微生物の増殖を抑制し、炎症反応(免疫応答)を促進させる作用がある。
発熱は、ウイルスなどの病原体の増殖を抑制したり、免疫(B細胞など)の活性を高める効果があると言う。ウイルスは、35~36℃の低い体温での方が増殖し易く、B細胞(Bリンパ球)は、38~39℃の高い体温の方が活躍し易い。
生物が、病原物質に対して、体温を上げて反応するのは、ヒトなど哺乳類に限らず、魚類、爬虫類、鳥類において認められる。魚類は、外因性の発熱物質が投与されると、体温を上げる為に、温かい水のほうに泳いで行く。爬虫類のトカゲは、外因性の発熱物質が投与されると、体温が上がるまで、日向に横たわるという。
健康な小児なら、39℃以下の発熱は、一般に治療を要しない。発熱し、体温が高くなると、患児は、不機嫌になったり、食欲が低下する。そのような状態が長引く場合、解熱剤(アセトアミノフェンなど)の投与により、機嫌や食欲が、改善する。解熱療法は(解熱剤を使用することは)、慢性の心肺疾患、代謝異常、神経疾患などを有するハイリスクの患者や、熱性痙攣を起こしやすい子には、有益とされる。解熱療法は(解熱剤を使用しても)、症状を軽減する以外には、感染症の経過を変えないと言う説もある。しかし、PGE2には、発熱作用や、疼痛(発痛増強作用)があるが、炎症性サイトカインの産生抑制作用(免疫抑制作用)や、細胞膜安定作用があるので、解熱療法により、PGE2の産生を抑制すると、却って、生体が、壊れ易くなるおそれがある。
注4:G蛋白質共役型受容体ファミリーには、G蛋白(α/β/γの3量体型)が共役している。
細胞外の受容体にリガンド(アセチルコリン、ホルモンなど)が結合すると、受容体の細胞質ゾル側の構造が変化し、G蛋白(GTP結合蛋白)と結合する。すると、G蛋白のαサブユニットがGTPと結合し、活性型αサブユニットとなり、βγ複合体と解離する。活性型αサブユニットは、2秒程度の後に、アデニル酸シクラーゼ(AC)系や、ホスホリパーゼC(PLC)系を活性化させる。活性型βγ複合体は、Kチャネルなどに結合して、チャネルを開放する。活性型αサブユニットが、結合しているGTPを加水分解して、GDP結合型の不活性型αサブユニットになると、βγ複合体と再び会合して、不活性型G蛋白に戻る。このようにG蛋白は、分子スウィッチ(分子スイッチ)として、機能している。
1).アデニル酸シクラーゼ系(AC系):
アデニル酸シクラーゼ系(AC系)では、ACにより、ATPからcAMPが生成され、cAMP依存性Aキナーゼ(protein kinase A:PKA)のCサブユニットを活性化させる。活性化されたAキナーゼのCサブユニットは、核内に移行して、標的遺伝子の転写活性を高め、様々な生理作用を示す。
Aキナーゼ(PKA)は、cAMP依存性キナーゼ。
G蛋白→アデニル酸シクラーゼ(AC)活性化→細胞内cAMP濃度増加→Aキナーゼ(PKA)活性化→Ca2+--ATPase活性化→細胞内Ca2+濃度の低下→血管平滑筋弛緩
あるいは、
G蛋白→アデニル酸シクラーゼ(AC)活性化→細胞内cAMP濃度増加→Aキナーゼ(PKA)活性化→蛋白質リン酸化→脱顆粒抑制(肥満細胞等)
なお、Aキナーゼ(PKA)は、cAMP依存性キナーゼで、ホルモン感受性リパーゼ(HSL)を活性させる。
2).ホスホリパーゼC系(PLC系):
ホスホリパーゼC系(PLC系)では、PLCにより、ホスファチジルイノシトール2リン酸(PIP2)が分解され、イノシトール3リン酸(IP3)とジアシルグリセロール(DAG)が生成される。
IP3(イノシトール3リン酸)は、小胞体のCa2+チャネル(IP3受容体)に結合して、Ca2+を細胞質ゾルに放出させたり、ミトコンドリアから、Ca2+を放出させ、細胞内Ca2+濃度を上昇させる。細胞膜のチャネルからも、Ca2+が流入する。Ca2+は、Ca2+-カルモジュリン依存性キナーゼ(CaMK)を活性化させ、標的蛋白質をリン酸化させる。
Ca2+は、Cキナーゼ(PKC:カルシウム依存性蛋白質リン酸化酵素)と結合する。Ca2+と結合したPKCは、活性化される。PKCはまた、細胞膜のDAGにより活性化される。活性化されたPKCは、標的蛋白質(セリン/スレオニン)をリン酸化させ、様々な細胞応答を示す。
G蛋白→ホスホリパーゼC→IP3生成→小胞体のCa2+チャネルからCa2+放出→細胞内Ca2+濃度の上昇→Ca2+がCキナーゼ(PKC)と結合→PKCが細胞膜にリクルートされ細胞膜のDAGにより活性化→標的蛋白質(セリン/スレオニン)をリン酸化
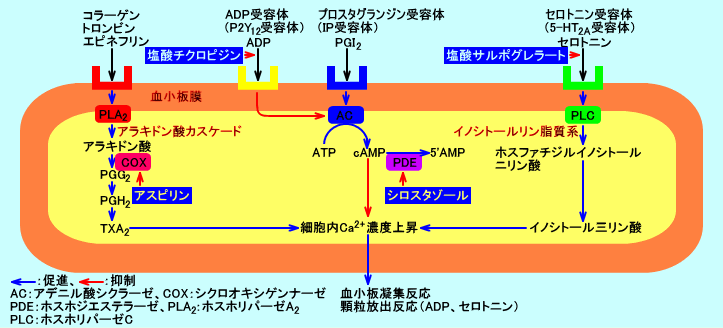
血管平滑筋には、ノルアドレナリン(ノルエピネフリン:NE)や、アンジオテンシンII(ATII)、エンドセリンなどに対する受容体(α1受容体等)が存在する。これらの受容体に、ノルエピネフリン(NE)などが結合すると、G蛋白のGαqに
GTPが結合し、ホスホリパーゼC(PLC)が活性化される。ホスホリパーゼC(PLC)により、PIP2が分解され、IP3と DAG が産生される。IP3は、小胞体のCa2+チャネルを開き、細胞内のCa2+濃度(Cac)を高める。細胞内Ca2+濃度(Cac)の上昇は、細胞膜上のCa2+チャネルを開口させ、細胞外からも細胞内に、Ca2+を流入させる。細胞内Ca2+濃度増加や、Cキナーゼ(PKC)が活性化される。PLCにより産生されたDAGも、PKCを活性化する。活性化されたPKCは、ミオシン軽鎖キナーゼ(MLCK)を活性化させ、血管平滑筋を収縮させる(血圧が上昇する)。
α1受容体-PLC-Ca2+↑-PKC:
皮膚、内臓などの血管平滑筋:ノルアドレナリン→α1受容体→G蛋白(Gαq)→ホスホリパーゼC(PLC)活性化→PIP2分解→IP3が小胞体のCa2+チャネルを開く→細胞内Ca2+濃度増加→Cキナーゼ(PKC)活性化→ミオシン軽鎖キナーゼ(MLCK)活性化→血管平滑筋収縮(昇圧作用)
血管平滑筋には、アドレナリン(エピネフリン:E)のβ2受容体(β2受容体)が、存在する。β2受容体は、アドレナリンが結合すると、刺激性G蛋白(Gαs蛋白)を介して、アデニル酸シクラーゼ(AC)を活性化する。アデニル酸シクラーゼ(AC)により生成されるcAMPは、Aキナーゼ(PKA)を活性化させる。Aキナーゼ(PKA)は、筋小胞体のCaポンプを活性化させ(Ca2+-ATPaseのリン酸化)、筋小胞体にCa2+を蓄積させ(強心作用、心拍増加作用)、細胞内Ca2+濃度(Cac)を、若干低下させる。また、Aキナーゼ(PKA)は、ミオシン軽鎖キナーゼ(MLCK)を不活性化(リン酸化)させ、アクチンを活性化させる(リン酸化されたミオシン軽鎖キナーゼは、Ca2+結合能が低下する)。このように、cAMPは、Aキナーゼ(PKA)を活性化させ、血管平滑筋を弛緩させる(冠動脈や、骨格筋の血管が拡張し、血流が増加する)。気管支は、β2受容体刺激により、拡張する。ノルアドレナリンは、β2受容体刺激作用が弱い。
β受容体-AC-cAMP↑-PKA-Ca2+↓:
冠動脈など:アドレナリン→β2受容体→刺激性G蛋白(Gαs蛋白)→アデニル酸シクラーゼ(AC)活性化→細胞内cAMP濃度増加→Aキナーゼ(PKA)活性化→ミオシン軽鎖キナーゼ(MLCK)不活性化→アクチン活性化→血管平滑筋弛緩(血流増加作用)
心筋:アドレナリン→β1受容体→刺激性G蛋白(Gαs蛋白)→アデニル酸シクラーゼ(AC)活性化→細胞内cAMP濃度増加→Aキナーゼ(PKA)活性化→Ca2+-ATPase活性化→筋小胞体にCa2+が蓄積(細胞内Ca2+濃度は若干低下)→筋収縮時に多量のCa2+が細胞内に放出される→筋収縮増強(強心作用、心拍数増加作用)
アドレナリンは、α2受容体を介しては、抑制性G蛋白(Gαi)を介して、アデニル酸シクラーゼ(AC)を抑制する。
アドレナリンの受容体には、α受容体(血管収縮による昇圧作用を現わす)と、β受容体(冠動脈や骨格筋の血管弛緩させ、血流を増加させ、筋小胞体にCa2+を蓄積させ、強心作用を現わす)とが存在する。
α1受容体(α1受容体)は、ホスホリパーゼC系(PLC系)を介して、細胞内Ca2+濃度を増加せ、Cキナーゼ(PKC)を活性化させ、血管平滑筋を収縮させる。
β2受容体(β2受容体)は、アデニル酸シクラーゼ系(AC系)を活性化させ、細胞内cAMP濃度を増加させ、Aキナーゼ(PKA)を活性化させ、細胞内Ca2+濃度を低下させ、血管平滑筋を弛緩させる。
α受容体は、アドレナリンとノルアドレナリンに反応する(結合する)が、 β2受容体は、アドレナリン(エピネフリン)により強く反応する。アドレナリンは、心拍数を増加させるが、ノルアドレナリンは、心拍数を減少させる(ノルアドレナリンのα1受容体刺激により、昇圧作用が現れると、圧受容器反射により、心拍数が減少し、昇圧を減じさせる)。アドレナリンは、総末梢抵抗を減少させ、ノルアドレナリンは、総末梢抵抗を増加させる(アドレナリン:β受容体刺激作用、ノルアドレナリン:α受容体刺激作用)。
ノルアドレナリンは、心臓以外の組織の血管(動脈)を収縮させる。アドレナリンは、骨格筋の血管を拡張させ(血流を増加させる)、他の組織の血管を収縮させる。
アドレナリンは、収縮期血圧を上昇させるが、拡張期血圧を低下させ、脈圧を広げる。アドレナリンによる血圧上昇(収縮期血圧上昇)によっては、圧受容器(圧受容体)反射は強くないので、アドレナリンによる強心作用の効果で、心拍数も、心拍出量も増加する。
アテコールアミン(ノルアドレナリンやアドレナリン)は、収縮期血圧を上昇させるが、ノルアドレナリンは、圧受容体による反射性徐脈の為、心拍出量を低下させる(末梢循環抵抗を増大させる)。アドレナリンは、心拍出量を増大させる(末梢血管抵抗を減少させる)。
| 作用 | アドレナリン | ノルアドレナリン |
| 心拍数 | ↑ | ↓ |
| 収縮期血圧 | ↑ | ↑↑ |
| 拡張期血圧 | ↓ | ↑ |
| 全末梢循環抵抗 | ↓ | ↑↑ |
| 心拍出量 | ↑ | ↓ |
α受容体は、カテコールアミン(ノルアドレナリンやアドレナリン)により、血管平滑筋を収縮させ、昇圧作用を示す。α受容体は、血小板凝集に関与する。
β受容体は、カテコールアミンにより、心臓に対する変力効果や変時効果により、強心作用や、心拍数増加作用を示す。β受容体は、冠動脈や、骨格筋の血管を拡張させ、血流増加作用により、代謝を促進させる。β受容体は、気管支拡張作用を示す。β受容体は、グリコーゲン分解や、脂肪分解(HSLによる)に関与する。
α1受容体(Gαq、Gαi)は、血管壁の血管平滑筋(postsynaptic)に存在し、血管平滑筋を収縮させる(昇圧作用により、全身の血圧が上昇し、昇圧作用を示す)。α1受容体は、末梢血管(動脈)を収縮させ、血圧を上昇させる。α1受容体は、β2受容体とは異なり、冠動脈や骨格筋の血管を収縮させる。
α2受容体(Gαi)は、ノルアドレナリン神経系(presynaptic)に存在し、ノルアドレナリン遊離を調節する。血小板に存在し、血小板凝集させる(アデニル酸シクラーゼを抑制する)。α2受容体は、交感神経からのノルアドレナリンの放出を抑制する(negative feedback)。
β1受容体(Gαs)は、心筋に存在し、心臓の収縮力を増強させる。腸にも存在する。β1受容体は、AC活性化→cAMP上昇→PKA活性化により、筋小胞体のCaポンプ(Ca2+-ATPase)を活性化させ、筋小胞体にCa2+を蓄積させ、強心作用を示す。また、洞房結節を刺激し、心拍数増加作用を示す。β1受容体は、強心作用や心拍数増加作用を現わす。β1受容体は、主に心臓を刺激し、β2受容体は、主に、末梢血管、気管支等を刺激する。
β2受容体(Gαs)は、平滑筋に存在し、気管支平滑筋や、骨格筋の血管平滑筋を弛緩させる。β2受容体は、心臓の冠動脈や、骨格筋の血管(血管平滑筋)に存在し、血管を拡張させる(血流増加作用を示す)。β2受容体は、冠動脈や、骨格筋の血管や、気管支等を拡張させる。
β3受容体は、脂肪細胞に存在し、脂肪分解を促進させる。
アドレナリン等は、(α1受容体の)Gαq蛋白を介して、PLC系(ホスホリパーゼC)を活性化させ、ジアシルグリセロール(DAG)を生成し、PKC(Cキナーゼ)を活性化させて、Na+/H+交換輸送体と、Na+-HCO3-共輸送系を活性化させる。AC系(アデニル酸シクラーゼ系)が活性化されると、細胞内cAMP濃度が上昇し、PKA(Aキナーゼ)が活性化され、Na+/K+-ATPase、H+/K+-ATPaseが活性化される。
アセチルコリン(Ach)やヒスタミンは、血管内皮細胞に作用し、NOを産生させ、すると NO が産生される。NOは、平滑筋のグアニル酸シクラーゼ(guanylate cyclase:GC)を活性化させ、cGMPを生成させ、Gキナーゼ(protein kinase G:PKG)を活性化させる。Gキナーゼ(PKG)は、リン酸化ミオシンを脱リン酸化させて、平滑筋を弛緩させる。
アデノシンは、心筋細胞のA1受容体を介しては、アデニル酸シクラーゼ(AC)を抑制し、cAMPを低下させ、酸素消費を抑制する(収縮力を抑制する)。
アデノシンは、冠血管(冠動脈)のA2受容体を介しては、アデニル酸シクラーゼ(AC)を活性化させ、cAMPを上昇させ、冠血管を拡張させる。
アデニル酸シクラーゼ(AC)系のアデニル酸シクラーゼ(AC)や、ホスホリパーゼC系のホスホリパーゼ(PLC)は、細胞膜に存在する。
アデニル酸シクラーゼ(AC)系で作用するAキナーゼ(PKA)は、細胞質内に存在する。
ホスホリパーゼC系で作用するCキナーゼ(PKC)は、細胞膜や細胞膜に存在する。cPKC(classical isoforms of PKC)は、Ca2+やDAGにより活性化される。nPKC(novel isoforms of PKC)は、Ca2+によっては活性化されず(calcium insensitive)、DAGにより活性化される。aPKC(atypical PKC)は、Ca2+やDAGにより活性化されない。
注5:アラキドン酸の含有量は、可食部100g当たり、豚もも肉28mg、鶏卵155mg、鯖(サバ:生)189mg、牛乳3mg。魚は、EPAも含んでいるが、同時に、魚は、アラキドン酸も多く含んでいる。
アラキドン酸の含有量は、脂肪酸総量100g当たり、豚脂身もも0.3g、鶏卵全生卵1.7g、さば生1.4g、牛乳普通牛乳0.1g。
参考文献
・吉野加津也、他:サイトカインと炎症 小児科 35:1233-1246, 1994.
・永倉俊和:抗アレルギー薬の使い方 小児科 36:921-932, 1995.
・市川陽一:RAの骨萎縮の機序は? Chronic Disease(臨牀消化器内科別冊) No.3, 118-120, 1990年.
・今井正、他:標準薬理学 第6版 (医学書院、2001年).
・大野岩男:薬剤性腎障害 日本醫事新報 No.4108(2003年1月18日)、21-27頁.
・田村充利、他:子宮内膜症の発症機序と病態 日本医師会雑誌 第134巻・第3号、375-378、2005年.
・藤井美穂:子宮内膜症の治療-疼痛管理を中心とした薬物療法 日本医師会雑誌 第134巻・第3号、388-392、2005年.
・斎藤英昭:外科的侵襲とプロスタグランジン NIKKEI MEDICAL(日経メディカル) 1995年6月15日号、109-110.
・荒川哲男、他:対談 マーロックス再考 第2回 まだまだ続く新知見 NIKKEI MEDICAL 1993年7月10日号、124-126.
・松井秀樹:Pemirolast potassiumおよびhydrocortisoneのヒト好酸球遊走に対する抑制効果 炎症(日本炎症学会雑誌) Vol.12 No.5, 467-473, 1992年.
・鹿取信:図説<炎症>シリーズ④ 炎症とその制御-薬理学の立場から- 日本医師会雑誌 昭和55年4月1月号.
・安保徹:医療が病いをつくる 免疫からの警鐘 岩波書店(2001年).
・安保徹:免疫革命 講談社インターナショナル、2003年.
・免疫調節と炎症調節 The Experiment & Therapy、No.580、2頁、1981年.
・古川漸、笹井敬子:小児膠原病の治療 プロスタグランディン 小児内科 Vol.17 No.3、423-428、1985年.
・松田幸次郎、他訳:医科生理学展望(原著6版、丸善株式会社 1975年).
・大川健太郎、中島重徳:最近の喘息治療薬の機序と役割(1) β遮断剤 医薬ジャーナル、31-37、1989年.
・半場道子:痛みのサイエンス 新潮選書(2004年).
・永倉俊和:抗アレルギー薬の使い方、小児科、Vol.36, No.8、921-931、1995年.
・高血圧診療のてびき 厚生省・日本医師会編、日本医師会雑誌 第104巻第2号、平成2年7月15日.
・市川陽一:RAの骨萎縮の機序は?、関節リウマチの診断と治療、臨床消化器内科 別冊、Chronic Disease、No.3 1990年、118-119頁.
・吉原重美:乳児のアレルギーと呼気性喘鳴、小児科診療、69(10) : 1453-1460, 2006.
・大柴晃洋:ロイコトリエン拮抗薬-抗アレルギー薬における位置づけ-、小児科、Vol.41 No.4、581-589頁.
・和泉孝志:PAF受容体、Medical Tribune、2007年7月5日、42頁.
・香川芳子:五訂食品成分表2005(女子栄養大学出版部、2005年).
・河野陽一:講座 呼吸器疾患の新治療 乳幼児用気管支喘息治療薬『モンテルカストナトリウム(シングレア)細粒4mg』、呼吸、26巻8号、741-746頁、2007年.