インスリン
【ポイント】
インスリンは、グリコーゲン合成を促進し、解糖を促進し、糖新生を抑制する。
インスリンは、肝臓での糖新生を抑制し、グリコーゲン合成を促進することで、肝静脈へのブドウ糖放出を抑制する。正常人では、肝臓での糖新生は、低濃度のインスリンでも抑制される。他方、肝臓でのブドウ糖取り込みは、インスリンの作用に依存しないGLUT2により行われる為、インスリン濃度で差が生じにくい(インスリンは、肝臓の糖放出率を低下させ、糖取り込み率を増加させる)。
インスリン(インシュリン:insulin)が作用するのは、主に、筋肉(骨格筋、心筋)、脂肪組織、肝臓。その内、骨格筋は、血糖の約70%を取り込む。
インスリンは、筋肉、脂肪組織では、グルコース(ブドウ糖)の細胞内への取り込みを促進し、また、肝臓では、肝静脈へのブドウ糖放出(糖新生)を抑制し、血糖値を低下させる。
インスリンは、筋肉(骨格筋、心筋)、脂肪組織で、糖輸送担体(GLUT4)の発現を増加させ、ブドウ糖(グルコース)の細胞内への取り込みを、10〜20倍、増加させる。
インスリンは、食事摂取後に、肝臓での糖放出(糖新生)を抑制し、骨格筋や肝臓での糖取り込みを増加させ、上昇する血糖値(血中グルコース濃度)を、低下させる。
運動中は、インスリン分泌が低下しているにも拘らず、骨格筋のAMPKが活性化され、GLUT4のトランスロケーションが起こり、運動筋へのグルコース取り込みが、行われ、血液中の糖(ブルコース)が、運動筋のエネルギー源として、利用される。
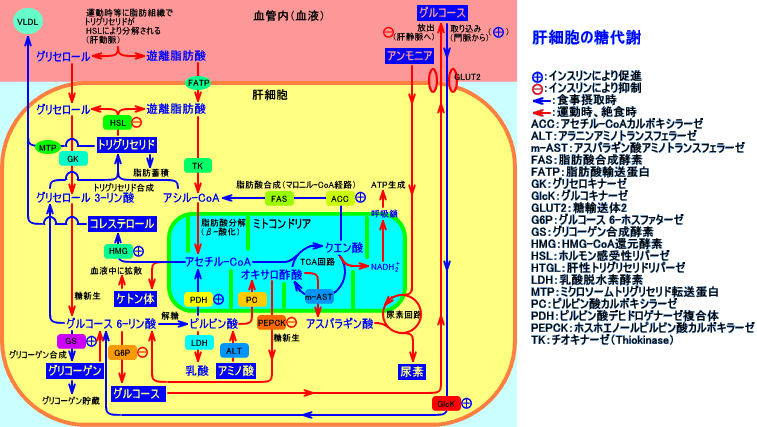
肝臓は、摂食時には、門脈中に追加分泌されたインスリンの作用で、瞬時に、グルコースの放出(糖放出)を抑制し、グルコースを取り込む(糖取り込み)。
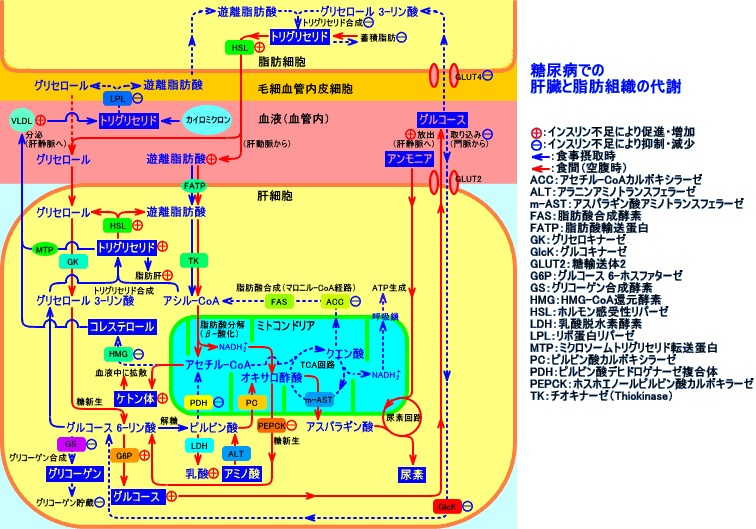
糖尿病では、肝臓の安静時の糖放出率(糖新生)が増加し、食後の糖取り込み率が低下している。
糖尿病では、空腹時には、糖新生抑制作用のあるインスリンが不足する為、肝臓での糖新生が亢進する(空腹時高血糖になる)。
糖尿病では、食後には、肝臓の(門脈血からの)糖取り込みや、筋肉や脂肪組織の糖取り込みが低下し、また、肝臓での糖新生が抑制されず、糖放出の抑制が低下する(食後高血糖になる)。
糖尿病では、細胞外(血液中など)のブドウ糖(グルコース)は、過多に存在し、細胞内にブドウ糖が取り込まれる。しかし、インスリンの作用が低下している為、脂肪酸やアミノ酸を代謝して、エネルギーを、まかなっている。また、糖尿病では、筋肉の蛋白質合成が低下し、血中のアミノ酸濃度が上昇し、肝臓でのアミノ酸からの糖新生が、亢進している。
インスリンの作用が不足すると、Na+/K+-ATPase(Na+,K+-ATPase)の活性が低下し、細胞内Na+濃度が増加し、高K血症を来たす:インスリンの作用が不足した糖尿病昏睡では、細胞内では、NaとHは増加し、Kは減少する
糖尿病で高血糖になるのは、インスリン不足の為、主に、肝臓での糖新生が抑制されないことが原因。
食事で摂取され、消化管から門脈血中に入ったグルコース(ブドウ糖)は、肝臓で取り込まれ、残りが、肝静脈から血中に入り、動脈血中に入ったグルコースは、主に、骨格筋のGLUT4により取り込まれる。
インスリンは、筋肉、脂肪組織で、細胞内へのグルコース取り込みを促進させ、グリコーゲン(筋細胞)や中性脂肪(脂肪細胞)として貯蔵させる。
インスリンは、グリコーゲン合成を促進し、解糖を促進し、糖新生を抑制する。
糖輸送担体(GLUT4)は、インスリンの標的臓器である、筋肉(骨格筋、心筋)、脂肪組織で、グルコースの取り込みを、促進させる:GLUT4は、通常は、細胞内小胞膜に局在していて、インスリン刺激により、細胞膜へ移動(トランスロケーション)され、細胞膜上に発現し、グルコースの細胞内への取り込みを、10〜20倍、増加させる。
運動は、インスリンと無関係に、糖輸送担体(GLUT4)の発現を増加させ、筋肉細胞内へのグルコース取り込みを、増加させる。
インスリンの作用が効きにくい糖尿病では、脂肪組織から遊離脂肪酸の放出が増加し、カイロミクロンやVLDLの異化が障害され、肝臓でのVLDL産生が増加し、LDLの生成が増加し、高脂血症(高FFA血症、高TG血症、高コレステロール血症)を、来たす。
インスリンの作用が不足すると、Na+/K+-ATPaseの活性が低下し、細胞内Na+濃度が増加し、高K血症を来たす:インスリンの作用が不足した糖尿病昏睡では、細胞内では、NaとHは増加し、Kは減少する。体内全体としては、Naは減少(腎臓からのNa再吸収の減少による)、Hは増加(代謝性アシドーシスによる)、Kは減少(腎臓からのK排泄の増加による)。
なお、糖尿病でも、インスリン抵抗性による、高インスリン血症がある段階では、インスリンにより交感神経系が刺激され、Na+/H+交換輸送体の活性(やNa+/K+-ATPaseの活性)が亢進し、尿細管からのNa+再吸収が増加して、体内のNa+量が増加して、高血圧を来たしやすい。
肝臓では、糖輸送担体(GLUT2)が、最初から細胞膜上に存在しているので、肝臓では、細胞内へのグルコース取り込みは、インスリンの作用に依存しない。
インスリンは、脂肪組織や肝臓で、中性脂肪の分解を抑制する(インスリンは、HSLを抑制する)。
肝臓は、必要時に、インスリンによって貯蔵したグリコーゲンの分解や、糖新生によって、グルコースを血液中に供給する。
脳神経細胞は、グルコースを主なエネルギー源にしているが、細胞内へのグルコース取り込みは、インスリンに依存しない。
血糖値(血液中のグルコース濃度)を下げる作用のあるホルモンは、インスリンだけ。
糖尿病では、細胞外(血液中など)のブドウ糖(グルコース)は、過多に存在し、細胞内にブドウ糖が取り込まれる。しかし、インスリンの作用が低下している為、脂肪酸やアミノ酸を代謝して、エネルギーを、まかなっている。また、糖尿病では、筋肉の蛋白質合成が低下し、血中のアミノ酸濃度が上昇し、肝臓でのアミノ酸からの糖新生が、亢進している(特に、インスリン分泌が欠乏した糖尿病では、肝臓での糖新生が、亢進する)。
糖尿病では、肝臓や骨格筋中のグリコーゲン含量は、いつも、低下している。糖尿病では、インスリン不足により、グルコースが、グルコキナーゼにより、グルコース 6-リン酸に変換されず、UDP-グルコースを経るグリコーゲン合成は、低下する。
糖尿病では、リポ蛋白リパーゼ(LPL)の活性が低下して、カイロミクロンやVLDLの異化が障害され、血液中の中性脂肪(トリグリセリド)が増加し、高TG血症(高中性脂肪血症=高トリグリセリド血症)になる。糖尿病では、LDL受容体の活性が低下し、高LDL血症(高コレステロール血症)になる。糖尿病では、ホルモン感受性リパーゼ(HSL)により脂肪分解が起こり、脂肪組織で中性脂肪が分解されて、遊離脂肪酸(FFA)とグリセロールが、血中に遊離され、高FFA血症になる。
インスリンが沢山分泌されると、脂肪細胞内に中性脂肪が蓄積し(肥満になる)、インスリンの分泌が不足する(インスリンの作用が低下する)と、血液中に中性脂肪が蓄積する(高中性脂肪血症になる)。
1.インスリンの作用
インスリンは、栄養素の同化を促進し、筋肉、脂肪組織、肝臓に取り込む。
インスリンが作用するのは、主に、筋肉(骨格筋、心筋)、脂肪組織、肝臓。この中で、骨格筋は、糖利用(グルコース利用)の約70%を占めるので、骨格筋にインスリン感受性がある(インスリン抵抗性でない)ことは、インスリンが血糖降下作用を現わす上で、重要。脂肪組織に取り込まれるグルコース(ブドウ糖)は、(血糖の)3%程度に過ぎないと言う。
1).糖質代謝
・グルコースの細胞内への取り込みを促進させる
インスリンは、グルコース(ブドウ糖)の細胞膜のGLUT4を介する、細胞内への取り込みを促進させる。
このインスリンの作用は、筋肉(特に、骨格筋)の筋肉細胞、脂肪組織の脂肪細胞で、起こる。インスリンは、脳(視床下部を除く)、腎尿細管(注1)、胃腸の細胞には、作用しない。
・グリコーゲン合成を促進させる
インスリンは、グリコーゲン合成酵素(glycogen synthase)を活性化させ、グリコーゲン合成を促進させる。
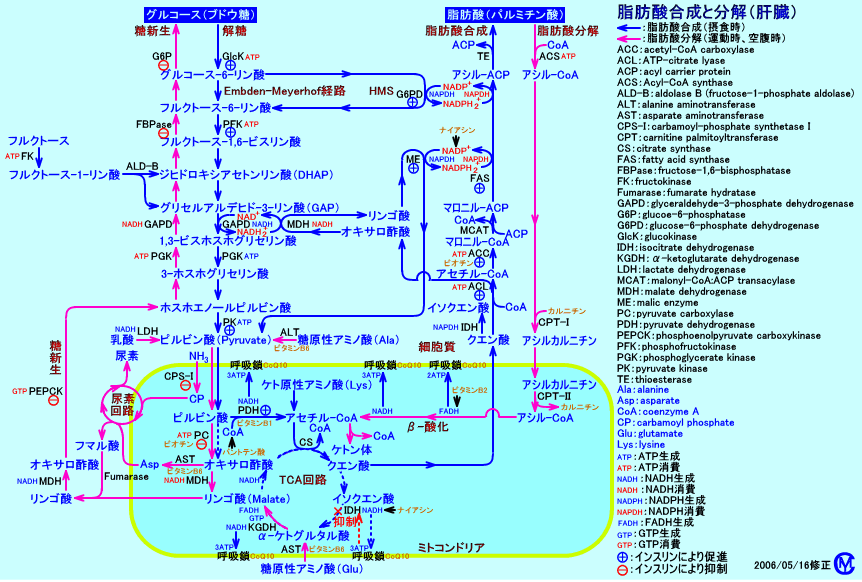
このインスリン作用は、肝臓で起こり、グリコーゲン分解を抑制し、アミノ酸、乳酸、グリセロールなどからの糖新生を抑制し、グルコース放出を抑制する。その結果、肝臓に、グリコーゲンが、貯蔵される。
・解糖系を促進し、糖新生を抑制する
インスリンは、解糖系の律速酵素(調節酵素)である、グルコキナーゼ、ホスホフルクトキナーゼ、ピルビン酸キナーゼ、ピルビン酸デヒドロキナーゼを誘導、あるいは、活性化させる。
また、ピルビン酸カルボキシラーゼ、PEPCK(注2)、グルコース-6-ホスファターゼ(glucose-6-phosphatase )の転写を抑制し、糖新生を抑制する。糖新生を抑制するには、グリコーゲン分解を抑制するより、多くのインスリンを必要とする。
インスリンは、骨格筋で、ヘキソキナーゼ(hexokinase II)を活性化させ、解糖系を促進する。
⇒インスリンにより、グルコースは、グリコーゲンとして筋肉や肝臓に貯蔵される。また、解糖系が促進され、中性脂肪として脂肪組織に貯蔵される。糖新生は、抑制される。追加インスリン分泌が低下する糖尿病では、筋肉や肝臓へのグリコーゲン貯蔵が滞り、また、脂肪組織への中性脂肪の貯蔵が滞る。
2).蛋白質代謝
・蛋白質合成を促進させる
インスリンは、骨格筋に作用して、アミノ酸やK+の細胞内への取り込みを増加させて、蛋白質合成を促進させ、蛋白質の異化を抑制する(注3)。
3).脂質代謝
・脂肪分解を抑制する
インスリンは、脂肪分解(lipolysis:中性脂肪の分解)を抑制する:インスリンは、脂肪組織、肝臓で、ホスホジエステラーゼ(PDE)を活性化させ、cAMPを5'AMPに異化させ、cAMP濃度を減少させ(注4)、ホルモン感受性リパーゼ(HSL)の活性を抑制する。
インスリンは、ホルモン感受性リパーゼ(HSL)の活性を抑制するが、インスリンは、リポ蛋白リパーゼ(LPL)の合成(活性)は、促進させる:インスリンは、脂肪細胞内のトリグリセリド(中性脂肪)の分解を抑制し、血漿中のリポ蛋白中のトリグリセリド(中性脂肪)の分解を促進し、脂肪酸を、脂肪細胞内に取り込む。
インスリンは、細胞膜表面のLDL受容体の活性を高める作用があると言われている。
・脂肪酸合成を促進する
インスリンは、アセチル-CoAカルボキシラーゼ(acetyl-CoA carboxylase:脂肪酸合成の律速酵素)、HMG-COA還元酵素(HMG-COA reductase:肝臓におけるコレステロール生成の律速酵素)を活性化させ、脂肪酸やコレステロールの合成を高める。インスリンは、HMG-CoAシンターゼ(HMGS)の活性をは阻害する。
インスリンは、アセチル-CoAカルボキシラーゼ(ACC)や脂肪酸合成酵素(fatty acid synthetase:FAS)の合成を促進させ、脂肪酸合成を促進する(インスリンは、中性脂肪や、コレステロール生成を促進し、VLDLの合成・分泌を増加させる)。なお、脂肪組織に取り込まれるグルコース(ブドウ糖)は、(血糖の)3%程度に過ぎないと言う。
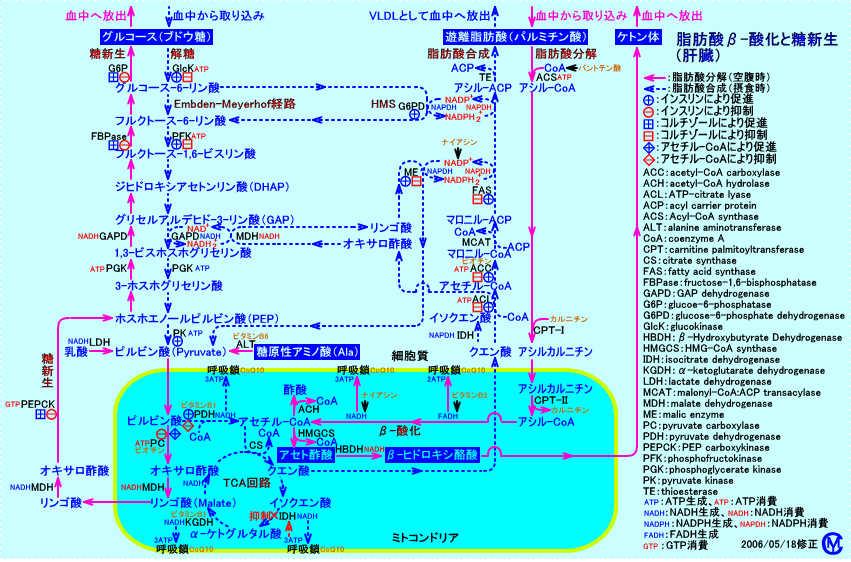
脂肪酸合成の中間代謝産物のマロニル-CoA(malonyl-CoA)は、カルニチンパルミトイルトランスフェラーゼ1型(CPT-1)活性を抑制し(アロステリック阻害)、脂肪酸分解(脂肪酸のβ-酸化)を抑制する。インスリンは、アセチル-CoAカルボキシラーゼ(ACC)の活性を促進し、脂肪酸合成を促進し、マロニル-CoA(malonyl-CoA)生成を促進する(脂肪酸分解を抑制する)。
⇒糖尿病で、インスリンが欠乏すると、ホルモン感受性リパーゼにより脂肪分解(lipolysis)が起こり、脂肪組織でトリグリセリド(中性脂肪)が分解されて、遊離脂肪酸(FFA)とグリセロールが、血中に遊離され、高FFA血症になり、体重は減少する。
糖尿病では、高VLDL血症に伴い、VLDLが異化されて生じるLDLの生成が増加し、また、LDL受容体の活性が低下し、高LDL血症(高コレステロール血症)になる。なお、小腸細胞内のACAT(acyl coenzyme A-cholesterol acyltransferase)活性が上昇し、腸管からのコレステロール吸収も、亢進することも、高コレステロール血症にさせる原因と考えられている。
糖尿病では、リポ蛋白リパーゼ(LPL)の酵素活性が低下し、カイロミクロンやVLDLの異化が障害され、血液中の中性脂肪(トリグリセリド)が増加し、高TG血症(高中性脂肪血症、高トリグリセリド血症)になる。
また、糖尿病で、リポ蛋白リパーゼ(LPL)の酵素活性が低下すると、VLDLが異化されて生成されるHDLも減少し、低HDL血症になる。なお、肝臓でのHDL合成が低下するのが、低HDL血症の原因とも言われている。
糖尿病に続発する高脂血症(二次性高脂血症)は、VLDLが増加するIV型と、VLDLとLDLとが増加するIIb型とか多いとされている(WHO分類)。重症の糖尿病では、V型の高脂血症になることがある。
肝臓では、細胞内と細胞外のグルコース濃度は、ほぼ、等しい:肝臓では、糖輸送担体(GLUT2)が、最初から細胞膜上に存在している。そのため、肝臓では、細胞内へのグルコース取り込み量は、インスリンの作用に依存しない。インスリンは、肝臓で、グルコキナーゼ(glucokinase)を活性化させ、間接的にグルコースの肝臓への取り込みを増加させる。肝臓は、糖新生により、グルコースを合成(新生)し、細胞膜のGLUT2を介して、血中に放出(供給)する。インスリンは、糖新生を抑制し、グリコーゲン合成を抑制することで、肝臓からのグルコース放出を抑制する(インスリンは、肝臓の糖放出率を低下させ、糖取り込み率を増加させる)。
糖尿病では、グルコースが細胞内へ取り込まれても、インスリン不足の為、解糖などの代謝が十分に行われない。その結果、エネルギー源は、脂肪酸に依存している:糖尿病では、飢餓状態と同様に、脂肪酸に、エネルギー源を依存している。
インスリンは、膵臓のランゲルハンス島のβ細胞で産生され、門脈血中に分泌され、肝臓や腎臓で、分解される。大量のインスリンが、肝臓と腎臓に結合している。インスリンは、脳や赤血球には、結合しない。
インスリンは、血漿蛋白のアルブミンやアポB蛋白(アポ蛋白B)を、増加させる。
2.インスリンは、GLUT4を介する細胞内へのグルコース取り込みを、促進させる
膵臓のラ氏島β細胞は、組織中のブドウ糖濃度に反応し、インスリンを分泌する。
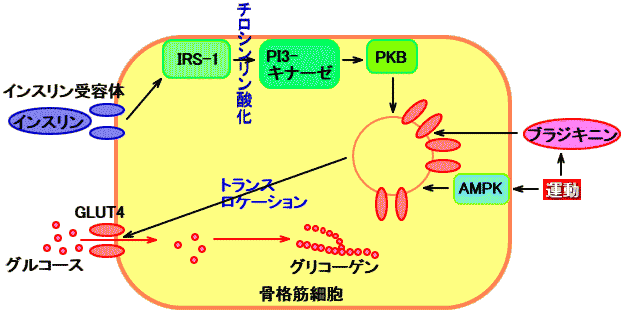
インスリンが、筋肉細胞や脂肪細胞に存在する、インスリン受容体に結合すると、チロシンキナーゼが活性化されて、IRS-1(insulin receptor substrate-1)などがチロシンリン酸化される。
IRS-1のリン酸化チロシンに、PI3-キナーゼ(phosphoinositide 3-kinase)、Grb2・Sos複合体、SHP-2が結合する。
PI3-キナーゼが活性化され、プロテインキナーゼB(PKB、Aktとも呼ばれる)が結合する。
PKBは、PDK1(phosphatidylinositol-dependent protein kinase 1)によりリン酸化され、活性型PKBになる。
活性化PKBは、細胞膜を離れ、種々の蛋白質をリン酸化する。
そのため、筋肉細胞(骨格筋、心筋)、脂肪細胞では、糖輸送担体(グルコーストランスポーター:glucose transporter)のGLUT4(グルットフォー:glucose transporter 4)含有小胞の細胞膜への移動(トランスロケーション:translocation)が促進され、細胞膜上にGLUT4が発現され、細胞内へのグルコース(ブドウ糖)の取り込みが促進される。
この時、K+も、細胞内に取り込まれる。
このように、GLUT4は、インスリン反応性組織(筋肉細胞、脂肪細胞)に発現し、インスリンに反応して、グルコース(ブドウ糖)とK+を、細胞内へ取り込む。
活性化PKB(Akt)は、インスリンの蛋白質合成の促進作用、グリコーゲン合成酵素の活性化作用にも関与している。
肝細胞では、グリコーゲン合成酵素が活性化され、グルコースからグリコーゲンが合成される。
取り込まれたグルコースは、グリコーゲンとして筋肉や肝臓に貯蔵ざれたり、解糖系を経て中性脂肪として脂肪組織に貯蔵される。
インスリン受容体の数は、インスリン濃度が増加すると減少する(down regulation)。
AMPキナーゼ(AMPK)は、運動により、筋収縮や虚血(Hypoxia)が起こり、骨格筋のATPが消費され、AMPやAMP/ATP比が上昇すると、活性化され、GLUT4のトランスロケーションを起こすとされる。
運動は、AMPキナーゼを活性化し、インスリンに依存せず、GLUT4のトランスロケーションを起こし、筋肉の細胞内へのグルコース取り込みを促進するので、糖尿病の治療に有用。
運動によって、骨格筋のAMPキナーゼが活性化されると、骨格筋は、糖や脂肪を取り込んで、燃やす。
運動中は、食後と異なり、インスリン分泌が低下しているにも拘らず、骨格筋のAMPKが活性化され、GLUT4のトランスロケーションが起こり、運動筋へのグルコース取り込みが、行われ、血液中の糖(ブルコース)が、運動筋のエネルギー源として、利用される。
運動療法は、AMPKを活性化させ、骨格筋細胞へのグルコース取り込みを促進させ、インスリン抵抗性を改善する。1回の運動時間は、10分以上が、望ましい。
アディポネクチン(注5)は、運動することなしに、AMPキナーゼを、活性化させ、インスリン抵抗性を、改善する。
運動は、ブラジキニンを筋肉から産生させ、GLUT4のトランスロケーションを起こし、細胞内へのグルコース取り込みを促進する。
有酸素運動を行うと、GLUT4の発現量が増加し、ミトコンドリア数も増加する。
図には示さないが、Grb2・Sos複合体は、Rasの活性化を経て、MAPキナーゼ(MAPK)を活性化させる。インスリン受容体に結合したインスリンは、PI3-キナーゼを介して、代謝作用(GLUT4の発現など)を現し、MAPキナーゼを介して、増殖作用を現す。
活性化されたPI3-キナーゼ(PI3K)は、グリコーゲン合成酵素を活性化させ、グリコーゲン合成を促進させる。
糖尿病では、インスリン抵抗性やインスリン分泌不足のため、筋肉や脂肪組織で、GLUT4を介する、細胞内へのグルコース取り込みが低下する(食後高血糖の要因になる)。しかし、空腹時高血糖になるのは、肝臓での糖新生が、亢進していことが要因とされる。
糖尿病で高血糖になるのは、肝臓での糖産生が増加する(糖新生とグリコーゲン分解の促進する)のが、主なる原因と言われる。
肝臓は、消化管で吸収され門脈を流れて来た外因性グルコースを積極的に取り込むが、末梢循環から肝動脈を経て再循環して来た内因性グルコースをは、僅かしか、取り込まない。他方、筋肉などの肝外組織は、末梢循環(動脈血)を流れる内因性グルコースを、良く取り込む。
3.糖輸送担体(GLUT)
糖輸送担体(グルコーストランスポーター:glucose transporter)は、GLUT(グルット)と、略される。
GLUT1は、殆んどの組織に発現(存在)している。
GLUT2は、肝臓と、膵臓のβ細胞に、発現している。
GLUT3は、小腸に発現している。
GLUT4は、心臓(心筋)、骨格筋、脂肪組織(脂肪細胞)に、発現する。インスリンの標的臓器である、筋肉(骨格筋、心筋)、脂肪組織には、GLUT4が、発現する。
GLUT5は、脳や、睾丸(testis)に発現している。
GLUTは、5種類存在するが、インスリンにより発現を調節されるのは、GLUT4。
肝臓以外の組織では、インスリンは、細胞膜のGLUT4の発現数を増加させ、グルコース(ブドウ糖)の細胞内への取り込み(uptake)を、増加させる。GLUT4は、予め細胞質内に予め形成されていて、細胞膜にリクルート(recruitment)され、細胞膜に発現する。
GLUT1は、脳や赤血球に存在する糖輸送担体で、インスリン非依存性にグルコース取り込みをする。
脳神経細胞は、グルコースを主なエネルギー源にしているが、細胞内へのグルコース取り込みは、インスリンに依存しない。
赤血球は、細胞膜上のGLUT1により、グルコースを取り込む。赤血球は、ミトコンドリアを有しないので、1分子のグルコースから、嫌気的解糖により、2分子のATPしか、生成出来ない。赤血球は、生成したATPを用いて、膜骨格を構成する蛋白質をリン酸化し、赤血球の形態と柔軟性(変形能)を維持する。
グルコース輸送担体GLUT1異常症では、髄液中の糖(髄液糖9が、低値を示す:髄液糖/血糖比0.4以下。GLUT1異常症では、赤血球への糖取り込みも、低下する。GLUT1異常症では、乳児期早期からの癲癇発作(てんかん発作)、失調、精神運動発達遅滞を認める。GLUT1異常症では、糖(グルコース)が脳内に十分に移行しないが、ケトン体は、移行するので、ケトン食により、癲癇、神経症状が、改善する。乳児期に発症し、精神運動発達遅滞、眼振、失調などを伴い、原因不明の癲癇(てんかん)で、症状が空腹時に増悪する症例は、GLUT1異常症の可能性があるので、空腹時髄液糖/血糖比を測定する。
GLUT1は、赤血球の膜蛋白の5%を占める。
GLUT2は、肝臓の肝細胞、膵臓のβ細胞、腎臓の近位尿細管細胞、小腸の上皮細胞の血管側に発現する糖輸送担体で、グルコース親和性は低い(Km:約20mM)が、最大輸送量(Vmax)は大きい。GLUT2は、濃度依存性に、グルコースと、ガラクトース(Gal)を、細胞内に取り込んだり、細胞外に放出する。
食事を摂取して、血糖値(血中のブドウ糖濃度)が上昇すると、膵臓のβ細胞で、GLUT2によりブドウ糖の取り込みが増加し、インスリンの分泌が増加する:膵臓のβ細胞では、GLUT2によりブドウ糖(グルコース)が細胞内に取り込まれ、グルコキナーゼ(glucokinase)により、グルコース-6-リン酸になる。グルコース-6-リン酸は、細胞外に拡散出来ない。グルコース-6-リン酸は、細胞質内(解糖系)や、ミトコンドリア内(TCA回路や呼吸鎖)で、代謝され、ATPが生成され、ATP/ADP比が上昇する。その結果、細胞膜のATP依存性Kチャネルが閉口して、脱分極が起こり、電位依存性Caチャネル(VDCC)が開口し、細胞外から細胞内にCa2+(カルシウムイオン)、Ca依存性のインスリン分泌(インスリン分泌顆粒の放出)が引き起こされる。
肝臓では、糖輸送担体(GLUT2)が、最初から細胞膜上に存在しているので、肝臓では、細胞内へのグルコース取り込みは、インスリンの作用に依存しない。インスリンは、肝臓で、グルコキナーゼ(glucokinase)を活性化させ、間接的にグルコースの肝臓への取り込みを増加させる。肝臓は、糖新生により、グルコースを合成(新生)し、細胞膜のGLUT2を介して、血中に放出(供給)する。糖新生の経路が肝臓に存在することにより、肝臓で、脂肪酸をβ-酸化して得られたエネルギーを、グルコースとして蓄積し、他の組織で、利用することが出来る。
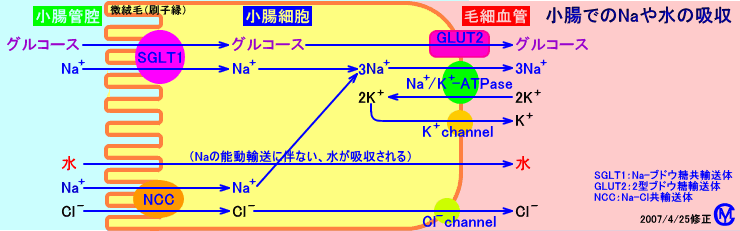
小腸や、腎臓の尿細管では、Na+は、グルコース(ブドウ糖)と共に、Na+-ブドウ糖共輸送体(SGLT:sodium-dependent glucose transporter)により、細胞内に取り込まれた後、Na+,K+-ATPaseにより、細胞外(血管側)に汲み出される。グルコースは、小腸では、GLUT2により、血管側に輸送される。
Fanconi-Bickel症候群は、GLUT2の欠損の為、肝型糖原病と、近位尿細管障害を伴なう。
GLUT2が欠損している為、肝細胞などの単糖類(グルコースやガラクトース)放出量が低下し、肝臓や腎臓にグリコーゲンが蓄積する。また、膵β細胞の血糖感受性が低下し、インスリン分泌が低下し、食後に、高血糖になる。
Fanconi-Bickel症候群は、新生児マススクリーニングで、総ガラクトースが、高値を示す。
GLUT4は、インスリン依存性に発現し、グルコース(ブドウ糖)取り込みを促進させる。GLUT4は、インスリンの標的臓器である、筋肉(骨格筋、心筋)、脂肪組織で、グルコースの取り込みを、促進させる:GLUT4は、通常は、細胞質(細胞内小胞膜)に局在していて、インスリン刺激により、細胞膜へ移動(トランスロケーション)され、細胞膜上に発現し、グルコースの細胞内への取り込みを、10〜20倍、増加させる。インスリン濃度が低くなると、GLUT4は、細胞膜から細胞質に、戻る。
運動は、インスリンと無関係にGLUT4の発現を増加させ、筋肉細胞内へのグルコース取り込みを、増加させる。
GLUT5は、消化管と腎臓に存在し、フルクトースを輸送する。
GLUT5は、小腸の微絨毛の吸収上皮細胞の刷子縁では、小腸管腔側にも、毛細血管側にも、存在する。
フルクトース(果糖:ショ糖がスクロースにより分解されても生成される)は、小腸管腔側のGLUT5(Na+非依存性グルコース輸送体)から、吸収上皮細胞に取り込まれ、毛細血管側のGLUT5から、毛細血管側の間質にに受動輸送(促通拡散)され、さらに、毛細血管内に単純拡散する。
4.インスリン抵抗性
インスリン抵抗性とは、インスリンが効き難く、血糖を低下させる為に、過剰にインスリンを分泌する状態のこと。
糖尿病の多くは、NIDDM(インスリン非依存型糖尿病、2型糖尿病)。
NIDDMは、インスリン抵抗性(インスリン感受性の低下)と、インスリン分泌不全が、要因となっている。
インスリン抵抗性(インスリンの作用低下)は、インスリンの標的組織である筋肉(骨格筋、心筋)、肝臓、脂肪組織で、見られる。
インスリン抵抗性は、脂肪細胞に脂質(中性脂肪)が蓄積した際に、それ以上、脂質が蓄積しないように、インスリン感受性を低下させ、細胞内へのグルコース取り込みを抑制する、生理的な防御反応と考えられる。
糖尿病の治療として、インスリン抵抗性を改善するためには、糖質摂取より、脂質摂取を控え、摂取カロリーを制限するのが、正道と思われる。
インスリン抵抗性になると、インスリンが過剰に分泌され、高インスリン血症になる。
高インスリン血症は、交感神経を活性化させたり、腎臓(近位尿細管、遠位尿細管、集合管)でNa+の再吸収を促進させて、高血圧の要因となる。高血圧になると、細胞内にNa+が流入しやすくなり、Na+/K+-ATPaseが働き過ぎて、十分働かなくなり、血管の中膜筋肉細胞などの細胞内に、Na+が蓄積する。Na+が蓄積すると、血管壁に水分が引き寄せられ、血管が肥厚し、血管内腔が狭くなり、血圧がさらに上昇する。
血中のアディポネクチン濃度の低下は、インスリン抵抗性と、非常に相関する。血中のアディポネクチン濃度の低下は、糖尿病の原因になるインスリン抵抗性の、良い指標となる。
高インスリン血症は、インスリン受容体を減少させる(down regulation)。
アンジオテンシンII(AII)は、インスリン抵抗性の一因。
TNF-αは、脂肪細胞がトリグリセリドを蓄積し大型化すると、(血中への)分泌量が増加する。
脂肪細胞が産生するTNF-αは、局所(脂肪組織のTNF-α濃度)のみならず、血中のTNF-α濃度を上昇させ、インスリン抵抗性を引き起こす。
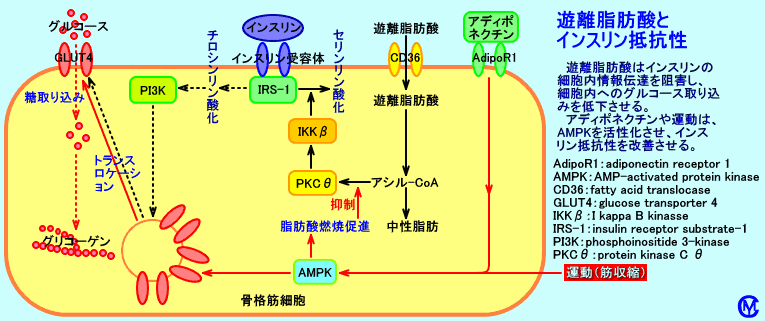
TNF-αは、骨格筋に作用して、インスリンによる、IRS-1のチロシンリン酸化やPI3-キナーゼの活性化を抑制し、GLUT4を介する、細胞内へのグルコース取り込みを抑制し、インスリンによるグリコーゲン合成を抑制する。
TNF-αは、脂肪細胞または筋肉細胞で、TNF-αの二つの受容体(TNFR1とTNFR2)の内、TNFR1を介して、スフィンゴミエリナーゼ(スフィンゴミエリンをセラミドにする酵素)を活性化させ、IRS-1を介して、インスリン受容体のリン酸化を抑制する。
TNF-αは、脂肪細胞からも分泌されるので、肥満者では、TNF-αの分泌が増加し、インスリン抵抗性が出現し易いと考えられている。
TNF-αは、IRS-1のみならず、糖輸送担体(GLUT4)の発現(転写)を抑制し、インスリン依存性の細胞内へのグルコース取り込みを抑制し、耐糖能に異常を来たさせ、糖尿病の発症に関連する。
遊離脂肪酸(FFA)も、脂肪細胞から産生される。血漿遊離脂肪酸(FFA)が上昇すると、筋肉細胞では、PI3-キナーゼが低下し、グルコースの取り込みが低下する(Randle効果)。
高脂肪食負荷や遊離脂肪酸(FFA)は、インスリン抵抗性を惹起させる。血中の脂質や遊離脂肪酸が増加し、骨格筋や肝臓に中性脂肪が蓄積すると、中性脂肪やアシル-CoA(脂肪酸-CoA)により、PKCθ(protein kinase C θ)、IKKβ(I kappa B kinase)が活性化され、IRS(骨格筋ではIRS-1、肝臓ではIRS-2)のセリンリン酸化が起こる。その結果、インスリンによるIRSのチロシンリン酸化やPI3-キナーゼ(PI3K)の活性化などが阻害され、GLUT4の細胞膜への発現が低下し、細胞内への糖取り込みが低下する(インスリン抵抗性)。ストレスによって、副腎髄質などからアドレナリンが分泌されると、脂肪組織でホルモン感受性リパーゼ(HSL)により中性脂肪が分解され、血中に遊離脂肪酸が増加する(ストレスはインスリン抵抗性を惹起する要因になる)。
肥満を解消すると、インスリン抵抗性が改善される。
インスリン抵抗性は、主に、脂肪細胞の肥大と相関する。
インスリン抵抗性は、NIDDM(2型糖尿病)の主要な要因である。
NIDDMのインスリン抵抗性は、脂肪細胞の肥大や、骨格筋細胞内の脂肪蓄積(intramyocellar lipid:IMCL)や、肝細胞内の脂肪蓄積(intrahepatic lipid:IHL)により、悪化するとされる。
食事療法や運動療法を行うと、体脂肪や内臓脂肪が減少し、インスリン抵抗性が改善し、インスリン感受性が高まる。このインスリン抵抗性の改善は、脂肪細胞の肥大の改善(脂肪細胞の大きさの減少)と相関する。しかし、インスリン抵抗性の改善は、骨格筋細胞内の脂肪蓄積(IMCL)の改善や、肝細胞内の脂肪蓄積(IHL)の改善とは、有意な相関がない。
各種降圧剤でも、ARB(アンジオテンシンII受容体拮抗剤)、ACE阻害剤、長時間作用型Ca拮抗剤、α遮断剤(α1ブロッカー)は、インスリン抵抗性を改善する(インスリン感受性を増加させる)。しかし、利尿剤、β遮断剤(βブロッカー)は、インスリン抵抗性を、増悪させるという(ただし、血管拡張性β遮断剤は、インスリン抵抗性を改善する)。(βブロッカー)の降圧作用は、プロスタグランジン(PG)が関与していて、インドメサシンによって、殆ど消失する。β遮断剤は、起立性低血圧(眩暈、動悸、失神等)を来たすことがあるので、少量より内服を開始し、漸増する。β遮断剤は、尿道平滑筋のα1受容体をも遮断して、尿が漏れ易くすることもある。β遮断剤は、心不全、気管支喘息、徐脈、II度以上の房室ブロック、心筋梗塞、レイノー症状のある場合には、投与してはならない(禁忌)。褐色細胞腫の患者には、α遮断剤で初期治療を行った後に、β遮断剤を投与する(褐色細胞腫の患者には、α遮断剤とβ遮断剤とを、併用する)。
Ca拮抗剤は、血清脂質に影響しなくても、抗炎症作用等により、動脈硬化を抑制する。
表 各種降圧剤のインスリン抵抗性や血清脂質への影響 降圧剤 インスリン抵抗性 LDL-コレステロール HDL-コレステロール トリグリセリド Ca拮抗剤(長時間作用型) ↓ → → → ACE阻害剤 ↓ → → → ARB ↓→ →↓ → → α遮断剤 ↓ ↓ ↑ ↓ β遮断剤 ↑ ↑ ↓ ↑ 利尿剤 ↑ ↑ ↓ ↑ ヒトは、1日約50単位のインスリンを必要とする。この量は、ヒトの膵臓内の貯蔵インスリン量の1/5に相当する。
グルコース(ブドウ糖)は、膵臓のβ細胞からのインスリン分泌を刺激する:この効果は、刺激後30〜60秒で、現れる。
マンノースや、程度は少ないがフルクトース(果糖)も、インスリン分泌(放出)を促進する。ガラクトース(Gal)、アラビノース(Arabinose)、キシロース(Xylose)は、インスリン分泌を促進しない。
アミノ酸の、アルギニンやリジンは、インスリン分泌を刺激すると言う。
糖尿病では、インスリンの作用や分泌が不足しているが、肝臓や全身の組織でのブドウ糖取り込み(血中からのグルコース消失速度)は、減少していない。糖尿病では、高血糖の為、ブドウ糖消費(血中からのグルコース消失速度)は、正常人より、むしろ多い(細胞内に取り込まれたブドウ糖の代謝は、インスリンの作用不足の為、低下し、アルドース還元酵素による還元が行われ、ソルビトールが蓄積する)。
脂質は、インスリン感受性を低下させる(インスリン抵抗性を増加させる)。
血中に遊離脂肪酸が増加すると、インスリンによる(筋肉や脂肪組織での)糖取り込みが、減少する。
HOMA-R(the homeostasis model assessment of insulin resisitance)は、インスリン抵抗性の指標として用いられる。
HOMA-R=空腹時血糖(mg/dL)×空腹時血中インスリン濃度(μU/mL)÷405
正常者では、HOMA-R=1(空腹時血糖=90mg/dL、空腹時血中インスリン4.5μU/mL)。
HOMA-Rは、血糖値やインスリン値が上昇すると、増大する。
QUICKI(quantiative insulin sensitivity check index)も、インスリン抵抗性の指標として用いられる。
QUICKI=1/[log(空腹時血中インスリン濃度)+log(空腹時血糖)]
5.ACE阻害剤によるインスリン抵抗性の改善
ACE阻害剤は、下記の機序により、インスリン抵抗性を改善する。
1).レニン・アンジオテンシン系(RA系)の阻害
アンジオテンシンII(AII)は、インスリンのインスリン受容体への結合による、IRS-1のチロシンリン酸化を抑制し、グルコースの骨格筋内への取り込みを抑制する。
ACE阻害剤は、AIIの産生を抑制し、インスリン抵抗性を改善する。
なお、AIIの受容体には、AT1とAT2が知られている。AT1受容体拮抗剤は、AIIのグルコースの骨格筋内への取り込み抑制作用を解除し、骨格筋へのグルコースの取り込みを促進し、インスリン抵抗性を改善する。しかし、AT1受容体拮抗剤は、下記のブラジキニンを介するインスリン抵抗性の改善効果は、期待出来ない。
2).ブラジキニン(BK)の分解抑制(ブラジキニンの増加)
ACE阻害剤は、ブラジキニンの分解を抑制する。
ブラジキニンは、PI3-キナーゼを介さずに、GLUT4の細胞膜上への移行(トランスロケーション)を促進させ、細胞内へのグルコース取り込みを促進させ、インスリン抵抗性を改善するとされる。
運動は、筋肉から、ブラジキニンの産生を増加させ、インスリン抵抗性を改善すると考えられる。
3).局所の血流増加
ACE阻害剤により、血管が拡張すると、血流が増加し、グルコース取り込みの機会が増える。
その他、ACE阻害剤は、IRS-1のチロシンリン酸化を抑制する、遊離脂肪酸やTNF-αの産生を抑制し、インスリン抵抗性を改善するという。
6.インスリンと高血圧
インスリンには、Na+/K+-ATPaseを、細胞質から、細胞膜にトランスロケーションさせる作用があるという。Na+,K+-ATPaseは、3ケのNa+を細胞外に汲み出すのに伴い、2ケのK+を細胞内に取り込む。
インスリンは、Na+,K+-ATPaseを、細胞膜に多く発現させ、その結果、細胞内Na濃度が減少したことが契機となって、Na+/H+交換輸送(尿細管で、原尿中から尿細管細胞内にNa+を取り込んで、H+を排泄する)が促進され、Na+の細胞内への受動輸送(再吸収)が促進されるものと思われる(注6)。
インスリン抵抗性のある糖尿病では、高インスリン血症の為、中枢神経系を介して、交感神経系が緊張し、血管が収縮し、血圧が上昇する(高血圧を来たす)。
また、(高血圧やインスリン抵抗性・不足の糖尿病では、)Na+,K+-ATPase活性が低下し、細胞内Na濃度が増加し、ミトコンドリアのNa+/Ca2+交換輸送体(ミトコンドリアにNa+を取り込んで、Ca2+を汲み出す)が刺激されるので、細胞内Ca2+濃度は、増加する。細胞内Ca2+濃度が増加することにより、細胞内pHが、アルカリに傾き、血管が収縮し、心筋や血管平滑筋の収縮性が増加するので、高血圧が増悪し易い。また、インスリンにより、(細胞膜の)Na+/H+交換輸送体が刺激され、細胞内pHが、アルカリに傾き、血管平滑筋が、増殖する。
インスリンは、NOを介して、血管を拡張させるという。
IL-1βは、膵臓のβ細胞からのインスリン分泌を、誘導型のNOS(iNOS)の発現を刺激して、抑制するという。
7.糖尿病性昏睡
糖尿病では、尿糖が増加するための浸透圧利尿と、インスリン作用不足により、水・電解質異常が生じ、糖尿病性昏睡の陥る危険がる。
糖尿病昏睡では、浸透圧利尿により、体内の水が喪失し、血漿中Na濃度が上昇し得るが、尿中にNaが喪失することと、血漿浸透圧(注7)が上昇して、細胞内から循環血液中に、水が移動し希釈される為、血漿中Na濃度は、必ずしも上昇しない。
インスリンは、Na+/K+-ATPaseを、細胞質から、細胞膜にトランスロケーションさせる作用がある。従って、インスリン作用不足は、Na+,K+-ATPaseによる、細胞内から細胞外へのNa汲み出しを、減少させる。また、腎臓でのNa再吸収が減少し、K+の排泄が増加する。その為、糖尿病性昏睡では、必ずしも、血漿中のNa濃度は、上昇しない。しかし、Kは、インスリン作用不足により、細胞内への取り込みが減少して、糖尿病性昏睡では、高K血症(高カリウム血症)を来たすことが多い。なお、インスリンの作用が不足すると、Na+/K+-ATPaseの活性が低下し、細胞内Kは、減少しているので、インスリン治療を開始すると、Na+,K+-ATPaseの活性が賦活化され、細胞内へのK取り込みが促進され、低K血症を来たし易いので、注意が必要。
代謝性アシドーシスでは、H+が、血液中から、細胞内に入り、交換に、K+とNa+が、細胞内から、細胞外(血液中)に出て、血清K濃度が上昇する。pHが0.1低下すると、血清K濃度は約0.6mEq/L上昇する。しかし、血清Na濃度は、元々、血清K濃度の30倍程度高いので、通常、有意に上昇しない。アルカローシスでは、逆に、H+が、細胞内から、血液中に出て、交換に、K+とNa+が、血液中から、細胞内に入り、血清K濃度が低下する。
このように、糖尿病性昏睡では、細胞内NaとHは増加し、細胞内Kは減少していることが多い。
糖尿病性昏睡には、高血糖高浸透圧性昏睡と、ケトアシドーシス昏睡(糖尿病性ケトーシスによる昏睡)と、乳酸アシドーシスによる昏睡がある。
高血糖高浸透圧性昏睡は、2型糖尿病の高齢者に、感冒などを契機に発症することが多く、血糖値が上昇して、血漿浸透圧は、350mOsm/L以上になり、極度に脱水する。
ケトアシドーシス昏睡は、1型糖尿病の若年者に、感染症などのストレスがかかることが誘因で、発症することが多く、血中pHは、7.3以下になる。高血糖を、治療で、急激に250mg/dl以下に下げると、脳浮腫が起こることがある。
メイロン(炭酸水素ナトリウム液)には、重炭酸イオン(HCO3-)が、7%液は0.833mEq/ml、8.4%液は1mEq/ml、含まれている。
糖尿病のケトアシドーシスの場合、重炭酸(メイロン)を投与すると、CNSアシドーシスを来たし、中枢神経系障害の原因となると言う。
糖尿病のケトアシドーシスの場合、点滴(輸液)とインスリン治療により、ケトン体が、減少すると、アシドーシスも改善する。糖尿病のケトアシドーシスの場合、炭酸水素ナトリウム(メイロン)を投与すると、血糖値が正常化するに従い、アルカローシスと低K血症が起こり、呼吸抑制が起こり、動脈血CO2濃度が上昇するおそれがあるので、糖尿病のケトアシドーシスの場合、原則として、炭酸水素ナトリウム(メイロン)は、投与しない。
乳酸アシドーシスによる昏睡は、糖尿病患者が、アルコール(注8)を多飲して起こることが多い。乳酸アシドーシスによる昏睡は、ビグアナイド薬(経口血糖降下剤)、サリチル酸なども、原因で起こる。乳酸アシドーシスによる昏睡は、意識障害(傾眠、昏睡)、消化器症状(嘔吐、腹痛)、ならびに過呼吸を呈し、多くの場合、ショック状態に陥る。
なお、糖尿病では、肝臓での乳酸産生の亢進、骨格筋での乳酸利用の低下、肝臓での糖新生系の亢進(肝臓でのアラニン取り込みの増加)、ミトコンドリア内でのピルビン酸脱水素酵素活性の低下(ピルビン酸や乳酸がアセチル-CoAに変換されない)などのため、乳酸が増加しやすい。
8.食後血糖値と肝臓
食後血糖値は、肝臓の糖取り込み率で、規定される。
インスリンは、肝臓で、グルコキナーゼ(glucokinase)を活性化させ、間接的にグルコースの肝臓への取り込みを増加させる。
2型糖尿病(NIDDM)では、食後のインスリン分泌が不足し、肝臓での糖取り込み率の増加が鈍く、また、肝臓での糖放出率の低下も鈍い為、食後高血糖になり易い。
糖尿病での食後高血糖は、肝臓での糖放出率の低下の遅滞や、糖取り込み率の低下が原因で、空腹時高血糖は、肝臓での糖放出率の増加(糖新生の亢進)が原因。
糖尿病では、肝臓の安静時の糖放出率(糖新生)が増加し、食後の糖取り込み率が低下している。
肝臓は、食後、門脈から流入するグルコース(消化管で吸収され門脈を流れて来た外因性グルコース)を積極的に取り込むが、肝動脈から流入するグルコース(末梢循環から肝動脈を経て再循環して来た内因性グルコース)をは、僅かしか、取り込まない(門脈シグナル)。
肝臓は、空腹時(絶食時)には、グルコースを放出(糖放出)し、摂食時(食後)には、グルコースを取り込む(糖取り込み)。摂食時に、インスリンは、肝臓からの糖放出を抑制する。
肝臓は、絶食時には、糖新生やグリコーゲン分解により、グルコースを放出(糖放出)する。しかし、肝臓は、摂食時には、門脈中に追加分泌されたインスリンの作用で、瞬時に、グルコースの放出(糖放出)を抑制し、グルコースを取り込む(糖取り込み)。
肝臓は、絶食時には、糖新生や、グリコーゲン分解(糖原分解)により、糖放出するが、肝臓は、食事摂取により、門脈から糖(グルコース)が流入して来ると、糖放出を瞬時に中止し、糖を積極的に取り込む。その結果、健常人では、食後、門脈に多量の糖が流れても、静脈中の血糖値は、140mg/dLを超えることがない。健常人では、門脈に流入した糖は、1回、肝臓を通過する度に、最低38%が、肝臓に取り込まれる。
食事で摂取され、消化管から門脈血中に入ったグルコース(ブドウ糖)は、肝臓で取り込まれ、残りが、肝静脈から血中に入り、動脈血中に入ったグルコースは、主に、骨格筋のGLUT4により取り込まれる。
2型糖尿病(NIDDM)では、食後、膵β細胞からインスリンが急峻に分泌されない為、肝臓での糖取り込み率の増加が鈍く、また、肝臓での糖放出率の低下も鈍い為、食後、高血糖になり易い。なお、肝臓での糖放出率の低下も鈍いのは、インスリン分泌が不足の為、インスリンによる糖新生の抑制が不十分であることと、インスリン抵抗性の為、脂肪組織での脂肪分解が亢進し、肝臓での糖新生の材料が豊富になることが、原因。
体重が正常な2型糖尿病(NIDDM)患者では、空腹時血糖値(FBG)が140mg/dlの場合、健常人と比して、肝臓の糖放出率が高い。肝臓の糖放出率が高い程、空腹時血糖値が、上昇する。
正常人では、肝臓でのグリコーゲン合成は、夕食後4時間でピークとなり、以後、減少する。
糖尿病では、肝臓のグリコーゲン貯蔵量は、正常人の約半分しかない。
糖尿病では、UDP-グルコースを経るグリコーゲン合成は低下するが、三炭糖を経て糖新生経路を経るグリコーゲン合成は、低下しない(インスリン不足により、グルコースが、グルコキナーゼにより、グルコース 6-リン酸に変換されず、UDP-グルコースを経るグリコーゲン合成は、低下する。三炭糖が、糖新生経路に入り、グルコース 6-リン酸に変換され行われるグリコーゲン合成は、低下しない)。
肝臓は、インスリンの作用に依存しないGLUT2により、ブドウ糖を取り込む。
正常人の肝臓での糖新生は、食後、低濃度のインスリンでも、高濃度のインスリンと同様に、抑制される。しかし、正常人の肝臓でのブドウ糖取り込みは、食後、高濃度のインスリンでないと、増加しない。正常人では、食後、肝臓の糖新生は、低濃度のインスリンでも抑制され、また、肝臓のブドウ糖取り込みは、インスリン濃度で差が生じにくい。
(インスリンが不足した)糖尿病患者の肝臓は、糖新生が亢進しているが、食後、肝臓でのブドウ糖取り込みは、行われている。
糖尿病患者の肝臓は、食後、高濃度のインスリンを(門脈に)注入しても、門脈血からのブドウ糖取り込みは、低下している。
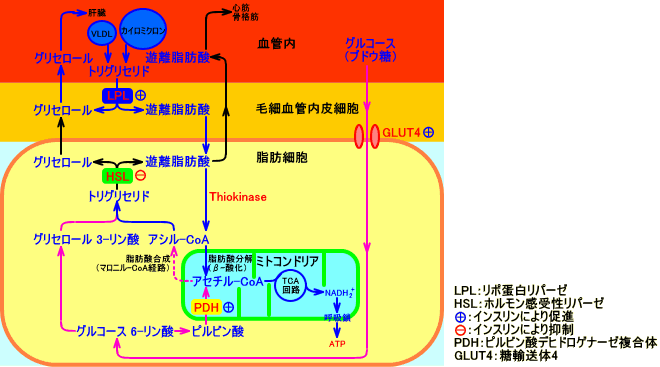
リポ蛋白リパーゼ(LPL)は、トリグリセリド(中性脂肪)を、脂肪酸とグリセロールに分解する。
LPLは、カイロミクロンやVLDLに含まれる、アポC-IIと言うアポ蛋白により、活性化され、カイロミクロンやVLDLのトリグリセリド(中性脂肪)を、脂肪酸とグリセロール(モノグリセライド)とに、加水分解する。
脂肪組織では、LPLにより分解した脂肪酸を、再び、トリグリセリドとして、貯蔵する。脂肪組織以外の組織(心筋や骨格筋など)では、LPLにより分解した脂肪酸を、エネルギー源として利用する。
インスリンは、脂肪組織のリポ蛋白リパーゼ(LPL)の活性を上昇させる:インスリンは、筋肉(特に心筋)のLPL活性をは、低下させる(注9)。なお、インスリンは、ホルモン感受性リパーゼ(HSL)の活性をは、抑制する。
インスリンは、脂肪細胞内のトリグリセリド(中性脂肪)の分解を抑制し、血漿中のリポ蛋白中のトリグリセリド(中性脂肪)の分解を促進し、脂肪酸を、脂肪組織(脂肪細胞)内に取り込む。脂肪組織(白色脂肪細胞)では、glycerokinase(グリセロールを、グリセロール-3-リン酸にする酵素)の活性が低いので、LPLによりトリグリセリドが分解される際に、脂肪酸と同時に放出されるグリセロールは、血漿中を、肝臓に輸送される。
脂肪細胞では、アシル-CoAと、グリセロール 3-リン酸(α-グリセロリン酸)から、トリグリセリドが生成される。グリセロール 3-リン酸は、インスリンにより脂肪細胞内に取り込まれるグルコース(ブドウ糖)の代謝(解糖)により、供給される。
糖尿病では、VLDL、カイロミクロンレムナント、VLDLレムナント(IDL)が増加する。また、LDLの異化で生成されるHDL(特に、HDL2)が、低下する。NIDDMでは、門脈における高インスリン血症が、肝臓での脂肪酸合成を増加させ、VLDL合成を促進させ、LDLが増加する。
血糖の上昇により、LDLのアポB蛋白のリジン残基が糖化され、LDL受容体(LDLレセプター)
への親和性が低下し、LDLが血中に増加し、動脈硬化の原因となる、酸化LDLも増加する。
糖尿病では、小腸上皮細胞で、コレステロールの吸収を促進させるアシル-CoA:コレステロールアシルトランスフェラーゼ(ACAT:acyl-CoA cholesterol acyltransferase)の活性が増加していて、コレステロールを含んだカイロミクロンレムナントが増加しやすい。
トリグリセリド(中性脂肪)は、グルコースの代謝(解糖)で生成されるグリセロール-3-リン酸代謝と遊離脂肪酸とから、合成される。
脂肪組織の脂肪細胞は、トリグリセリドが過剰に蓄積すると、トリグリセリド合成をしない為、インスリン抵抗性になり、トリグリセリドの原料となるグルコースや、脂肪細胞を取り込まず、LPLによる血中のトリグリセリド(カイロミクロンやVLDL)の分解も、行われない。その為、血中には、グルコース、遊離脂肪酸、トリグリセリドが増加する(高血糖、高中性脂肪血症になる)。
糖尿病(NIDDM)では、肝臓への遊離脂肪酸やグルコースの流入が増加する(肝臓では、グルコースの取り込みは、GLUT2により行われるので、インスリンの作用に依存しない)。その結果、糖尿病では、肝臓でのトリグリセリド(中性脂肪)の合成が増加する(増す持って高中性脂肪血症になる)。さらに、アポ蛋白のアポBの合成も増加する。その結果、トリグリセリドを多く含むVLDLの粒子数が、血中に増加する。このVLDLは、LPL活性が低下している為、LPLによりLDLに異化されるより、直接、細胞へ取り込まれて、異化される。
10.1型糖尿病とウイルス感染
1型糖尿病では、自己抗体として、抗GAD抗体(抗GAD65抗体)が、陽性になる。
グルタミン酸脱炭酸酵素(Glutamic acid decarboxylase:GAD)は、グルタミン酸よりGABA(ガンマ-アミノ酪酸)を合成する際に作用する酵素。グルタミン酸脱炭酸酵素(GAD)は、主に、脳や膵ランゲルハンス島β細胞に存在する。
グルタミン酸脱炭酸酵素(GAD:グルタミン酸デカルボキシラーゼ)には、二つのアイソフォーム(GAD65とGAD67)が存在する。ヒト膵臓のランゲルハンス島細胞(特にβ細胞)には、GAD65が局在している。GAD65は、生後に発現し、抑制性の神経伝達物質であるGABAを増加させ、脳機能を正常に機能させる。GAD67は、胎児期から発現して、脳内でのGABA合成や口蓋形成に関与する。
グルタミン酸脱炭酸酵素(GAD:グルタミン酸デカルボキシラーゼ)には、二つのアイソフォーム(GAD65とGAD67)が存在する。ヒト膵臓のランゲルハンス島細胞(特にβ細胞)には、GAD65が局在している。
グルタミン酸脱炭酸酵素(GAD)は、コクサッキーB4ウイルスの構造蛋白(P2-C蛋白)と、相同性がある。マウスに、コクサッキーB4ウイルスを感染させると、抗P2-C蛋白抗体が出現し、次いで、抗GAD抗体が出現する。このことから、コクサッキーB4ウイルスに感染すると、抗P2-C蛋白抗体が産生され、交差免疫反応で、グルタミン酸脱炭酸酵素(GAD)を発現した膵β細胞を障害し、1型糖尿病(IDDM)を発症するとする説がある。コクサッキーB群ウイルスは、エンテロウイルス属のウイルスだが、心筋細胞、横紋筋細胞、リンパ球系細胞、膵細胞に、持続感染することが知られている。エンテロウイルスは、感染した宿主の細胞を障害するが、感染細胞膜上にウイルス由来の抗原(membrane antigen)は現れないので、ウイルス感染細胞を特異的に障害するキラーT細胞(細胞障害性の細胞性免疫)は、誘導されない。エンテロウイルス感染症では、中和抗体など、液性免疫が、主に、誘導される。エンテロウイルス感染症は、夏季に多く、“夏かぜ”として、上気道炎(鼻汁や咳嗽などのカタル症状に乏しい)、発熱、頭痛、筋肉痛、消化器症状(嘔吐や下痢)、発疹(皮疹)、粘膜疹などを呈する。エンテロウイルスの約1/3は、赤血球凝集素(HA)を有している。
急性発症しして死亡した1型糖尿病患者では、膵臓の膵島(ランゲルハンス島)に、組織障害性のCD8陽性Tリンパ球(CD8陽性キラーT細胞)が浸潤し、膵島炎により、β細胞が破壊されていることが報告されている。
プロウイルス(レトロウイルスの遺伝子が、宿主細胞のゲノムに組み込まれた状態)が、発現し、自己免疫疾患として、1型糖尿病を引き起こすとも言われる。1型糖尿病患者の膵臓のランゲルハンス島を培養して、レトロウイルスが検出されたと言う報告もある。
牛乳蛋白(Cow's milk protein)が、1型糖尿病発症の環境因子とする説もあったが、最近は、否定的とされる。牛乳蛋白でも、ウシアルブミン(bovine serum albumin:BSA)の配列が、膵臓の膵島蛋白(ICA69)の配列と、一部、相同性があり、牛乳を飲んで、ウシアルブミンに対する抗体が産生されると、膵島蛋白(ICA69)に対して、交差免疫を起こして、膵島を障害して、1型糖尿病が発症すると言う説も言われて来た。また、牛乳に含まれる、ラクトグロブリン、カゼインA1が、1型糖尿病の発症に関連していると言う説も存在した。
なお、牛乳でも、低温殺菌(66℃、30分)を飲んでいて、1型糖尿病を発症した成人例がある(コクサッキーB4ウイルスなどのエンテロウイルスは、エンベロープを有しないので、消毒薬への抵抗性が、強いが、56℃以上の加熱で、急速に不活化される。しかし、Ca2+やMg2+など二価イオンが存在すると、加温によるウイルス不活化が防がれると言う)。
牛乳の摂取量と1型糖尿病の発症には、高い相関関係が認められている。
(新生児に)母乳でなく、牛乳を飲ませて育てると、1型糖尿病の発症率が高くなる(日本では、新生児は、母乳かミルクで育てるが、西洋では、牛乳で育てるほうが多いと言う)。
牛乳で育てると、1型糖尿病発症の相対危険率が、1.6倍高くなると言う報告もある。
デンマークでは、1型糖尿病のハイリスクグループ(1型糖尿病患者の第一度近親者)の新生児には、生後6カ月間は、乳製品を含めて、牛乳由来の蛋白を摂取させないことにより、1型糖尿病の発症を抑制出来るか、検討されている(Cow's milk avoidance trial)。
1型糖尿病の発症率(15歳未満)は、フィンランド、スウェーデン、カナダ、ノルウェー、英国、ニュージーランドなどの国の方が、高い。
1型糖尿病の発症率が高いフィンランドやイタリヤのサルディニア島では、1型糖尿病の発症率(15歳未満)は、年間35〜40人/10万人(年間10万人当たり35〜40人)と言われる。それに比し、日本(北海道)では、1型糖尿病の発症率は、年間2〜3人/10万人と、少ない。
1型糖尿病の発症率を、世界九カ国で調査すると、牛乳消費量が一番少ない日本が、1型糖尿病の発症率が一番低かった(注10)。
11.インスリンと血小板凝集や血液凝固
インスリン抵抗性により糖尿病になると、動脈硬化になり、外因系血液凝固に関与する組織因子(TF)の発現が増加する(血管内で、血液が凝固し、血栓が形成され易くなる)。
インスリン非依存性糖尿病(2型糖尿病)の患者は、血液凝固因子の第VII因子、フィブリノゲン(フィブリノーゲン)が増加し、線溶系(線溶能)が低下している。
インスリン非依存性糖尿病(2型糖尿病)、肥満、高トリグリセリド血症(高中性脂肪血症)では、血液中のPAI-1の蛋白量(抗原量)と活性が、増加する(線溶系が抑制され、動脈硬化で血栓を形成され易い)。血液中のPAI-1が増加するのは、肝臓でのPAI-1産生の増加、内臓脂肪によるPAI-1産生と放出の増加によると考えられている。メタボリックシンドロームのように、内臓脂肪が増加すると、脂肪細胞からアディポネクチンや、TNF-α(高血糖を促進させる)や、アンジオテンシノーゲン(高血圧を来たす)や、PAI-1(血栓形成を促進させる)などが、分泌される。
糖尿病患者の血小板は、正常人に比して、活性化され易い:糖尿病患者の血小板は、血小板凝集能(ADP、トロンビンなど)が亢進し、自然に血小板凝集が起こり易い。また、フォスフォリバーゼC(ホスホリパーゼC:PLC)やフォスフォリバーゼA2(ホスホリパーゼA2:PLA2)の活性が上昇し、血小板の膜蛋白GPIIb/IIIaやGPIbα/V/VIの発現が増加し、フィブリノゲン結合が上昇している。
12.インスリンとケトン体
インスリンは、ケトン体を抑制する:脂肪細胞からの遊離脂肪酸の放出を抑制(HSLを抑制)し、肝細胞のミトコンドリア内への遊離脂肪酸(アシル-CoA)の取り込みを抑制(マロニル-CoAを取り込むCPT-Iを抑制)し、肝細胞のHMG-CoA合成酵素(HMGS)を抑制し、ケトン体産生を抑制する。食後など、インスリン分泌が増加すると、ケトン体産生が抑制される。
カテコールアミン(アドレナリンなど)やグルカゴンは、ケトン体産生を促進する:カテコールアミンは、脂肪細胞からの遊離脂肪酸の放出を促進(HSLを促進)し、グルカゴンは肝細胞のミトコンドリア内への遊離脂肪酸の取り込みや肝細胞のHMG-CoA合成酵素を促進し、ケトン体産生を促進する。飢餓(絶食)、発熱、ストレス状態(精神的、肉体的)は、グルカゴンやカテコールアミンの産生を亢進させ、ケトン体の産生が刺激される。
13.脂肪毒性と糖尿病
遊離脂肪酸は、脂肪毒性(adipotoxicity:細胞毒性)がある。
メタボリックシンドロームなどで、内臓脂肪から遊離脂肪酸が過剰に放出され、肝臓、筋肉(骨格筋)、膵臓(膵β細胞)など非脂肪組織に過剰に流入し、インスリン作用やインスリン分泌を阻害し、糖・脂質代謝異常を来たすと考えられている。
遊離脂肪酸によるインスリン分泌障害の程度は、膵ランゲルハンス島の中性脂肪(トリグリセリド)蓄積量と、良く相関すると言われる。
膵臓のβ細胞容量は、遊離脂肪酸によるアポトーシス(リポアポトーシス)により、減少する。
狭義の脂肪毒性:内臓肥満に伴い内臓脂肪から遊離脂肪酸が過剰に放出され、膵臓のβ細胞を障害し、インスリン分泌を阻害する
広義の脂肪毒性:放出された遊離脂肪酸が、肝臓、筋肉、脂肪組織など、インスリンが作用する臓器(組織)で、インスリン作用を阻害する(インスリン抵抗性が高まる)
遊離脂肪酸は、臓器で代謝や中性脂肪合成に利用されないと、短期的には内臓脂肪に蓄積し、長期的には皮下脂肪に蓄積する。
脂肪を過剰に摂取すると、血中インスリン濃度が上昇しても、全身組織(骨格筋など)での糖取り込みが減少する。
健常者に脂肪製剤を48時間静脈注射すると、インスリン感受性が低下し、インスリンが過剰に分泌され、高血糖になる(インスリン抵抗性が亢進する)。
骨格筋では、遊離脂肪酸輸送蛋白(FATP1)を不活化させると、高脂肪食でも、インスリン抵抗性が生じなくなる。アディポネクチンは、骨格筋では、遊離脂肪酸輸送蛋白(FATP)を活性化させ、遊離脂肪酸の分解(β-酸化)を促進させ、インスリン感受性を高めている(インスリン抵抗性を解除する)。
脂肪細胞では、遊離脂肪酸結合蛋白(fatty acis binding proteins:FABP)のap2、mal1を欠損させると、過食をしても、肥満やインスリン抵抗性が現れない。
(糖質より)脂肪摂取の過剰が、インスリン抵抗性を亢進させ、II型糖尿病を招くと考えられる。
おまけ
血糖自己測定する際に、微量採血する為の穿刺針(採血用穿刺器具)が用いられている。
このような採血用穿刺針の内、針のみがディスポーザル(使い捨て)で、穿刺針をセットする器具に、検査の度に、新しい穿刺針をセットする採血用穿刺器具だと、一人の患者を穿刺した際に、器具の穿刺針に面した箇所などに、血液が付着し、次に、別の患者を穿刺した際に、B型肝炎などを感染させてしまうおそれがある。
従って、一つ採血用穿刺器具を、複数の人に、共用して使用する場合(例えば、CRPなどを検査する為に、複数の患者に用いる場合)、器具全体がディスポーザルタイプである器具(フィンガーピットなど)を使用する方が、安全である。少なくとも、針の周辺部分が、ディスポーザルタイプである器具を、用いた方が、感染予防に良い。
注1:インスリンは、近位尿細管での、尿酸の再吸収を促進すると言う。
高インスリン血症がある糖尿病の患者では、高尿酸血症になる。
注2: PEPCK(phosphoenolpyruvate carboxykinase:[EC 4.1.1.31])は、糖新生の際に、オキサロ酢酸をホスホエノールピルビン酸にする際の、重要な律速酵素。
インスリンは、PEPCKの発現を抑制し、糖新生を抑制する。
ステロイド受容体ファミリーのPPARγ(peroxisome proliferator-activated receptor γ)は、PEPCKの転写を促進し、糖新生を促進させる。
糖尿病改善薬(経口血糖降下薬)のチアゾリジン誘導体(thiazolidine-2,4-dione)は、PPARγと結合する。
PPARγ欠損マウスは、高脂肪食下でも、インスリン感受性が、保持されている。
注3:インスリンは、BCAAの骨格筋への取り込みを促進するが、脳内の神経伝達物質の前駆体になる、芳香族アミノ酸(aromatic amino acids:AAA)の取り込みには、あまり影響しない。
注4:細胞では、グルコース濃度が低下すると、cAMP濃度は、上昇する。
注5:アディポネクチンは、レプチンと同様に、脂肪細胞から分泌されるホルモン(アディポカイン)で、インスリン抵抗性を改善する。
肥満や糖尿病で、アディポネクチンの血中濃度が低下すると、インスリン抵抗性が、増悪する。
アディポネクチンは、AMPキナーゼやPPARαを活性化させる。
アディポネクチンは、肝臓、骨格筋に、作用し、AMPキナーゼを活性化させる。AMPキナーゼが活性化されると、肝臓では、糖新生が抑制され、骨格筋では、糖(グルコース)の取り込みが増加し、血糖が、低下する。
PPARγ欠損マウスの小型脂肪細胞は、レプチンや、アディポネクチンが、多く発現され、また、分泌もされている。
アディポネクチンは、PPARαも、活性化する。AMPキナーゼも、PPARαも、脂肪を燃やす作用があり、β-酸化を促進し、筋肉や肝臓に蓄積した脂肪を、燃やし、中性脂肪の蓄積を抑制し、インスリン感受性を、高めるという。
アディポネクチンは、炎症性の(血管)内膜肥厚を抑制し、動脈硬化を抑制する。
日本人の約40%は、アディポネクチンを分泌しにくい、遺伝子多型(SNP)を有している。
食事療法で、体重を2〜3kg減少させたり、運動療法で、体重を減少させると、血中のアディポネクチンは、上昇する。
チアゾリジン誘導体は、肥大した大型脂肪細胞を、アディポネクチンを多く分泌する小型脂肪細胞に置換し、アディポネクチンの血中濃度を、2〜3倍程度に、上昇させる。チアゾリジン誘導体は、また、アディポネクチンの遺伝子の転写因子であるPPARγを活性化させ、PPARγは、アディポネクチンの遺伝子に結合し、その転写を促進させ、アディポネクチンの血中濃度を、上昇させる。
インスリンは、アディポネクチンの分泌を、急性に増加させる。
肥満のモデル動物では、アディポネクチンの分泌が低下するのみならず、筋肉や脂肪細胞で、アディポネクチン受容体が減少している(ダウンレギュレーション)。
アディポネクチン受容体は、酵母にも存在する。生命機構の進化の段階で、アディポネクチンは、飢餓の際に、筋肉や肝臓などで、脂肪の燃焼(β酸化)を促進させのに必要なホルモンとして、備えられたホルモンとも、考えられている。
アディポネクチンは、骨格筋では、遊離脂肪酸輸送蛋白(FATP)を活性化させ、遊離脂肪酸の分解(β-酸化)を促進させ、インスリン感受性を高めている。
アディポネクチンは、脂肪細胞(脂肪組織)から、特異的に分泌される。
II型糖尿病モデルマウスを用いた実験結果では、高脂肪食による肥満は、アディポネクチンの分泌を低下させる。また、アディポネクチンを補充すると、インスリン抵抗性や血清中の中性脂肪値が改善することが見出された。
アディポネクチンは、肝臓や筋肉で、脂肪の燃焼を促進する(AMPキナーゼを活性化し、肝臓では糖新生を抑制し、骨格筋では糖の取り込みを促進し、脂肪を燃焼する)。肝臓や筋肉に脂肪が蓄積すると、アディポネクチンが分泌されなくなり、脂肪が燃焼されなくなり、インスリン抵抗性などが現れる。
アディポネクチン受容体には、AdipoR1(肝臓、骨格筋な多数の組織に発現している)とAdipoR2(主に肝臓に発現している)の二種類が存在する。アディポネクチンは、AdipoR1に結合するとAMPキナーゼ経路(AMPK経路)を活性化させ、AdipoR2に結合するとPPARα経路を活性化させ、インスリン抵抗性などを改善する。
アディポネクチン受容体(AdipoR1とAdipoR2)が欠損したマウスを用いた実験結果では、肝細胞にアディポネクチンが結合せず、アディポネクチンにより血糖が降下しない。アディポネクチン受容体が欠損したマウスは、インスリン抵抗性と耐糖能障害を来たす。
肥満マウスでは、アディポネクチン受容体(AdipoR1とAdipoR2)は、正常(通常)の半分程度に減少している。
オスモチン(植物由来ペプチド)は、立体構造がアディポネクチンに類似している。酵母のオスモチン受容体は、動物のアディポネクチン受容体に類似している(相同体)。オスモチンは、野菜や果物に含まれている。
アディポネクチンは、遺伝子の相違により、脂肪細胞から分泌され易いタイプと、分泌され難いタイプの2種類存在する。日本人の約4割の人は、分泌され難いタイプのアディポネクチンを産生していて、血中アディポネクチン濃度は、分泌され易いタイプのアディポネクチンを産生している人に比して、2/3程度の量しかない。
アディポネクチンは、脂肪細胞に最も豊富に発現している。
アディポネクチンは、肝臓と骨格筋において、インスリン感受性を亢進させる。アディポネクチンは、肝臓と骨格筋でAMPキナーゼを活性化させ、肝臓で糖新生を抑制し、骨格筋で糖取り込みを促進させる。
アディポネクチンは、肝臓と骨格筋でPPARα活性化作用を示し、AMPキナーゼ活性化作用と共に、脂肪酸分解(脂肪酸燃焼)を促進し、トリグリセリド量(中性脂肪量)を低下させ、インスリン抵抗性を改善する。
アディポネクチンやアディポネクチン受容体は、アンジオテンシン経路(RAA系)の抑制(ARBなどによる)によって、増加する。
PPARαは、フィブラート系薬剤によって活性化される。
PPARγは、減量、チアゾリジン誘導体などにより活性化される。
肥満症では、アディポネクチン(Ad)産生や、アディポネクチン受容体(AdipoR)の発現が低下し、インスリン抵抗性が亢進し、メタボリックシンドロームに陥る。AMPキナーゼを活性化させたり、PPARαを活性化させることが、治療に有用と考えられている。
脂肪組織において、PPARα作動薬はアディポネクチン受容体(AdipoR)を増加させ、PPARγ作動薬は高分子量のアディポネクチン(HMW-Ad)を増加させる。
チアゾリジン誘導体は、PPARγ作動薬として、アディポネクチン依存性とアディポネクチン非依存性の経路を介して、抗糖尿病作用を発揮する。
注6:インスリンが、Na+/H+交換輸送体(Na+/H+チャネル)を活性化させ、Na+が細胞内に受動輸送され(細胞内Naが増加する)、その結果、Na+,K+-ATPaseがトランスロケーションにより、細胞膜に多く発現し、細胞内Na濃度が減少するとする説もあるようだ。
インスリンは、(赤)血球、筋、腎尿細管など、全身の細胞膜での電解質輸送を修飾する。インスリンは、Na+,K+-ATPase(Naポンプ)を活性化し、細胞内Na濃度を減少させる。他方、インスリンは、Na+/H+交換輸送体(Naを再吸収する)、Na+/Ca2+交換輸送体、Na+-アミノ酸共軛輸送体を活性化(促進)し、細胞内Na濃度を増加させる。
本態性高血圧患者の赤血球では、(高血圧の結果として、)Na+,K+-ATPase活性が低下している。Na+,K+-ATPase活性が低下すると、細胞内Na濃度が増加し、(細胞膜の)Na+/Ca2+交換輸送(Na+を取り込んで、Ca2+を汲み出す)を抑制するので、細胞内Ca2+濃度は、増加する。また、電位依存性Caチャネル(VDCC)が活性化され、細胞内Ca2+濃度が、増加する。細胞内Ca2+濃度の増加により、心筋や血管平滑筋の収縮性が増加し、高血圧を発症させる(血圧がさらに上昇する)。
インスリン依存性の血管拡張反応は、アデノシンや、Na+,K+-ATPaseの阻害剤で、減弱する(インスリンは、Na+,K+-ATPaseを活性化させ、細胞内Na+濃度を減少させ、ミトコンドリアのNa+/Ca2+交換輸送体を抑制し、細胞内Ca2+濃度の上昇を抑制する)。
注7:血糖値が高いと、血漿浸透圧が、上昇する。
血漿浸透圧(血清浸透圧)は、血漿中のNa濃度、K濃度、血糖値、BUN(尿素窒素)値で決定する。
血漿浸透圧=(Na+K)×2+血糖(mg/dL)÷18+BUN(mg/dL)÷2.8
血清K濃度は、Na濃度に比して著しく低値であり、血清Ca濃度、血清P濃度と同様に、血清浸透圧を規定する主な要素ではない。
そこで、血清浸透圧は、血清中のNa濃度、血糖値、BUN値から、下記の血清浸透圧の概算式を用いて、概算出来る。
血清浸透圧≒血清Na(mEq/L)×1.86+血糖値(mg/dL)÷18+BUN(mg/dL)÷2.8
なお、1%ブドウ糖液(1,000mg/dL)は、浸透圧は、約55mOsm/L。
5%ブドウ糖液は、浸透圧は、約277mOsm/Lで、pHは5程度(グルコース液の最も安定なpHは、3〜4と言われるが、注射用の5%ブドウ糖液は、滅菌に伴ない着色が生じない為に、pH3.5〜6.5に、調節されている)。
50%ブドウ糖液は、pHは3.5と酸性だが、滴定酸度(pH7.4にするのに要するNaOH量)は0.61と小さいので、血液中では、容易に緩衝される。
注8:元来、糖尿病患者では、肝臓の乳酸の取り込みが、亢進していたり、糖新生系が、亢進している。しかし、アラニンからのピルビン酸産生増加や、ピルビン酸脱水素酵素(PDH)の活性の低下などのため、糖尿病患者では、血中乳酸値が、上昇しやすい。
糖尿病患者が、アルコールを多飲して、乳酸アシドーシスによる昏睡を、起こすことが多いのは、アルコールにより、肝臓での乳酸利用が、低下し、高乳酸血症を来たすためとされる。
なお、アルコールは、糖尿病患者では、低血糖を誘発させる。アルコールの代謝は、主に、肝細胞内に局在するアルコール脱水素酵素(ADH)によって行われれ、その際、NAD+が、NADH2+に、還元される。参考文献では、「アルコールは、肝臓でのNADH/NAD+比を増加させ、phosphoenol pyruvate carboxylase(PEPC)の活性が低下することで肝糖産生(糖新生)が減少して低血糖を誘発する」と、説明されていた。しかし、細胞質ゾルのNADH/NAD+比が増加すれば、糖新生が亢進すると考えられる。むしろ、アルコールが、ADHによって代謝されて生成されるアセトアルデヒドが、肝臓のミトコンドリアを障害し、ミトコンドリア内での脂肪酸のβ-酸化が障害され、細胞質ゾルでのNADH/NAD+比が、低下して、糖新生が減少し、低血糖が、誘発されるのではないかと、考えられる。
なお、糖尿病患者が低血糖を起すのは、長時間の空腹、糖尿病患者への抗血糖剤やインスリンの投与が原因のことが多い。低血糖症状には、交感神経症状と交感神経症状の2種類がある。交感神経症状は、血糖が急激に低下した時(急性低血糖:血糖値35mg以下)に発生しやすく、頻脈、血圧上昇、冷汗などが現れる。副交感神経症状は、血糖がゆっくり低下した時(慢性低血糖:血糖値50mg以下)に発生しやすく、眠気、空腹感、異常行動などの症状が現れる。
注9:砂糖と脂肪を一緒に摂るのは、健康に良くない:砂糖の摂取で、インスリンが分泌され、脂肪組織のLPL活性が上昇する。脂肪摂取で、血液中に中性脂肪(カイロミクロン)が増加し、LPL活性が高まった脂肪組織に、取り込まれる。
砂糖(蔗糖)は、分解されると、ブドウ糖と果糖になる。果糖や砂糖を取り過ぎると、肝臓での果糖の代謝により、中性脂肪が多く合成され、脂肪肝や、高脂血症(高VLDL血症)の原因になる。
注10:1型糖尿病の発症率が、非常に、国や地域によって、差があることから、1型糖尿病の発症に関与する、環境因子乃至、遺伝因子格差が存在すると、考えられている。
1型糖尿病が、自己免疫により、膵臓のランゲルハンス島のβ細胞が破壊され起こることは確かだが、牛乳その物が、1型糖尿病の原因ではないと考えられる。牛乳(低温殺菌牛乳)に混在する混在する微生物(コクサッキーウイルスなど)や、牛乳に含まれる脂質(ホモジナイズされた脂肪球などによるアジュバント効果)が、自己免疫を引き起こしている可能性は、考えられる。
なお、牛乳の殺菌は、日本では、超高温短時間殺菌法(UHT:120〜130度で2秒殺菌)が一般的に用いられているが、世界では、高温短時間法(HTST:72度以上で15秒以上殺菌)が一般的に用いられている。低温殺菌牛乳は、低温保持殺菌法(LTLT:62〜65度で30分殺菌)で製造される。
参考文献
・ハーパー・生化学(丸善株式会社発行、1975年).
・ヴォート生化学(東京化学同人、2003年、第4刷).
・鈴木紘一、他:ホートン生化学 第3版(東京化学同人、2005年、第3刷).
・期待されるアディポネクチン DITN 第319号 2004年10月5日発行.
・片山茂裕:高血圧合併糖尿病の成因と病態 日本医師会雑誌 特別号 第130巻・第8号 S180-183, 2003年.
・河盛隆造:肝臓におけるインスリンの作用 日本医師会雑誌 第123巻・第9号 IS-05〜IS-08、2000年.
・河盛隆造:今日の血糖コントロールの考え方 日本醫事新報 No.4193(2004年9月4日)、1-5頁.
・小林正:骨格筋・脂肪細胞におけるインスリン作用 日本医師会雑誌 第123巻・第11号 IS-09〜IS-12、2000年.
・吉田俊秀、他:インスリンの作用と肥満 日本医師会雑誌 第124巻・第5号 IS-21〜IS-24、2000年.
・肝疾患診療マニュアル、日本医師会雑誌特別号 Vol.122、No.8、1999年.
・小林淳二、他:糖尿病−その予防と治療 糖尿病と脂質代謝異常(1) 日本医師会雑誌 第121巻・第1号 TY-37〜TY-40, 1999年.
・門脇孝:糖尿病はなぜ起こるか−成因と病態 日本医師会雑誌 116: 1395-1400, 1996年.
・矢崎義雄、他:Vascular Biologyと冠動脈疾患の治療(日経メディカル開発、1996年).
・松田幸次郎、他訳:医科生理学展望(原著6版、丸善株式会社 1975年).
・内山聖:説苑 インスリン抵抗性と高血圧 日本小児科学会雑誌 97巻6号、1345-1348, 1993年.
・丸浜喜亮:脂質パラメータのみかた 5.遊離脂肪酸 臨床医 vol.8, no.9, 22-23, 1982年.(Shrago, et al. : Comparative aspects of lipogenesis in mammalian tissues. Metabolism, 20 : 54, 1971.)
・村勢敏郎:今日の医学 2.リポ蛋白リパーゼ vol.8, no.9, 58-60, 1982年.
・押田芳治:Metabolic syndromeの運動療法 日本醫事新報 N0.4242(2005年8月13日)、12-16頁.
・清島満:インスリンによるリポ蛋白リパーゼの制御(質疑応答) 日本醫事新報 No.4236(2005年7月2日)、91-92頁.
・菊池方利:肝における糖調節機構について 日本醫事新報 No.4184(2004年7月3日)、21-30頁.
・泉井亮、金田研司:カラー図解 人体の正常構造と機能 IV肝・胆・脾、日本医事新報社、2003年.
・坂井建雄、河原克雄:カラー図解 人体の正常構造と機能 III消化管、日本医事新報社、2003年.
・春日雅人:インスリン作用と糖尿病発症機序の解明 日本医師会雑誌 第134巻・第10号別冊、59-63、2006年.
・中川友紀子、他:新生児マススクリーニングで発見され、重度の低血糖を示したFanconi-Bickel症候群の1例 日本小児科学会 107巻5号、769-774、2003年.
・遠藤文夫:血糖調節の分子機構 小児内科 Vol.22 No.2、11-16、1990年.
・一色玄:インスリンの作用機序−生化学的過程を中心に− 小児内科 Vol.17 No.12、21-26、1985年.
・水島裕、宮本昭正:今日の治療薬 解説と便覧 '97 (南江堂、1997年).
・貴田嘉一:小児における糖尿病の治療、糖尿病診療マニュアル、日本医師会雑誌、特別号、Vol.130 No.8、S202-S207、2003年.
・山田研太郎:急性合併症のための検査と処置、糖尿病診療マニュアル、日本医師会雑誌、特別号、Vol.130 No.8、S230-S233、2003年.
・松浦信夫:説苑 小児期発症インスリン依存性糖尿病の病因と遺伝疫学 日本小児科学会雑誌 99巻9号、1551-1554、1995年.
・田嶼尚子:小児糖尿病に関する最近の動向、日本醫事新報、No.4259、2005年12月10日、1-16頁.
・小林哲郎:1型糖尿病とその予防、日本醫事新報、No.4229、2005年5月14日、1-27頁.
・親宮正久:エンテロウイルス属−分類とその特徴、小児内科、Vol.21 No.5、11-15頁、1989年.
・斎藤康:インスリン抵抗性の判定方法、日本医事新報、No.4286(2006年6月17日)、90-91頁.
・厚生労働省医薬食品局:医薬品・医療器具等安全情報、No.224、採血用穿刺器具(針の周辺部分がディスポーザブルタイプでないもの)の取り扱いについて、日本医師会雑誌、第135巻・第4号、976-978頁、平成18年(2006年)7月.
・池田康夫:インスリンと血小板・凝固、日本医師会雑誌、第125巻・第5号、IS-45-IS-48頁、2001年3月(平成13年3月).
・秦葭哉:高脂血症とインスリン、日本医師会雑誌、第112巻・第1号、47-52頁、平成6年7月1日.
・小林哲郎、田中昌一郎:抗グルタミン酸デカルボキシラーゼ(GAD)抗体精密測定、最新 臨床検査のABC、日本医師会雑誌、第135巻・特別号(2)、生涯教育シリーズ−70、S280-S280、平成18年(2006年)10月.
・新宮正久:エンテロウイルス属−分類とその特徴、小児内科、Vol.21 No.5、1989-5、11-15頁.
・森島恒雄:エンテロウイルスの分子生物学、小児内科、Vol.21 No.5、1989-5、16-20頁.
・渡辺悌吉:いわゆる夏かぜとエンテロウイルス感染症、小児内科、Vol.21 No.5、1989-5、37-41頁.
・河合忠:目で見る初期診療の検査計画と結果の読み方(エスアールエル、1997年).
・上野隆登、依田道夫、森田恭代:肥満と肝疾患の関係について、日本医事新報、No.4334(2007年5月19日)、57-61頁.
・柏木厚典:メタボリックシンドローム Over View、日本医師会雑誌、第136巻・特別号(1)、メタボリックシンドローム up to date、S2-S13頁.
・島袋充生:脂肪毒性、日本医師会雑誌、第136巻・特別号(1)、メタボリックシンドローム up to date、S100-S103頁.
・山内敏正、原一雄、門脇孝:アディポネクチン、日本医師会雑誌、第136巻・特別号(1)、メタボリックシンドローム up to date、S179-S184頁.
・浅原実郎:アンチエイジング医学の基礎と臨床、日本抗加齢医学会専門医・指導士認定委員会、株式会社メジカルビュー社、2004年第1版第1刷発行(2006年第1版第8刷).
・Alicia Nadal, Pedro F Marrero, and Diego Haro: Down-regulation of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by insulin: the role of the forkhead transcription factor FKHRL1. Biochem J. 2002 August 15; 366(Pt 1): 289-297.